методы исследования структуры вещества по распределению в пространстве и интенсивностям рассеянного на анализируемом объекте рентгеновского излучения. Р. с. а. наряду с нейтронографией (См. Нейтронография) и электронографией (См. Электронография) является дифракционным структурным методом; в его основе лежит взаимодействие рентгеновского излучения с электронами вещества, в результате которого возникает Дифракция рентгеновских лучей. Дифракционная картина зависит от длины волны используемых рентгеновских лучей (См. Рентгеновские лучи) и строения объекта. Для исследования атомной структуры применяют излучение с длиной волны Рентгеновский структурный анализ1 Å, т. е. порядка размеров атомов. Методами Р. с. а. изучают металлы, сплавы, минералы, неорганические и органические соединения, полимеры, аморфные материалы, жидкости и газы, молекулы белков, нуклеиновых кислот и т.д. Наиболее успешно Р. с. а. применяют для установления атомной структуры кристаллических тел. Это обусловлено тем, что Кристаллы обладают строгой периодичностью строения и представляют собой созданную самой природой дифракционную решётку для рентгеновских лучей.
Историческая справка. Дифракция рентгеновских лучей на кристаллах была открыта в 1912 немецкими физиками М. Лауэ, В. Фридрихом и П. Книппингом. Направив узкий пучок рентгеновских лучей на неподвижный кристалл, они зарегистрировали на помещенной за кристаллом фотопластинке дифракционную картину, которая состояла из большого числа закономерно расположенных пятен. Каждое пятно — след дифракционного луча, рассеянного кристаллом. Рентгенограмма, полученная таким методом, носит название лауэграммы (См. Лауэграмма) (рис. 1).
Разработанная Лауэ теория дифракции рентгеновских лучей на кристаллах позволила связать длину волны λ излучения, параметры элементарной ячейки кристалла а, b, с (см. Кристаллическая решётка), углы падающего (α0, β0, γ0) и дифракционного (α, β, γ) лучей соотношениями:
a (cosα— cosα0) = hλ,
b (cosβ — cosβ0) = kλ, (1)
c (cosγ — cosγ0) =lλ,
где h, k, I — целые числа (Миллеровские индексы). Для возникновения дифракционного луча необходимо выполнение приведённых условий Лауэ [уравнений (1)], которые требуют, чтобы в параллельных лучах разность хода между лучами, рассеянными атомами, отвечающими соседним узлам решётки, были равны целому числу длин волн.
В 1913 У. Л. Брэгг и одновременно с ним Г. В. Вульф предложили более наглядную трактовку возникновения дифракционных лучей в кристалле. Они показали, что любой из дифракционных лучей можно рассматривать как отражение падающего луча от одной из систем кристаллографических плоскостей (дифракционное отражение, см. Брэгга - Вульфа условие). В том же году У. Г. и У. Л. Брэгги впервые исследовали атомные структуры простейших кристаллов с помощью рентгеновских дифракционных методов. В 1916 П. Дебай и немецкий физик П. Шеррер предложили использовать дифракцию рентгеновских лучей для исследования структуры поликристаллических материалов. В 1938 французский кристаллограф А. Гинье разработал метод рентгеновского малоуглового рассеяния для исследования формы и размеров неоднородностей в веществе.
Применимость Р. с. а. к исследованию широкого класса веществ, производственная необходимость этих исследований стимулировали развитие методов расшифровки структур. В 1934 американский физик А. Патерсон предложил исследовать строение веществ с помощью функции межатомных векторов (функции Патерсона). Американские учёные Д. Харкер, Дж. Каспер (1948), У. Захариасен, Д. Сейр и английский учёный В. Кокрен (1952) заложили основы так называемых прямых методов определения кристаллических структур. Большой вклад в развитие патерсоновских и прямых методов Р. с. а. внесли Н. В. Белов, Г. С. Жданов, А. И. Китайгородский, Б. К. Вайнштейн, М. Порай-Кошиц (СССР), Л. Полинг, П. Эвальд, М. Бюргер, Дж. Карле, Г. Хауптман (США), М. Вульфсон (Великобритания) и др. Работы по исследованию пространственной структуры белка, начатые в Англии Дж. Берналом (30-е гг.) и успешно продолженные Дж. Кендрю, М. Перуцем, Д. Кроуфут-Ходжкин и др., сыграли исключительно важную роль в становлении молекулярной биологии (См. Молекулярная биология). В 1953 Дж. Уотсон и Ф. Крик предложили модель молекулы дезоксирибонуклеиновой кислоты (ДНК), которая хорошо согласовалась с результатами рентгенографических исследований ДНК, полученными М. Уилкинсом.
В 50-х гг. начали бурно развиваться методы Р. с. а. с использованием ЭВМ в технике эксперимента и при обработке рентгеновской дифракционной информации.
Экспериментальные методы Р. с. а. Для создания условий дифракции и регистрации излучения служат рентгеновские камеры (См. Рентгеновская камера) и рентгеновские дифрактометры (См. Рентгеновский дифрактометр). Рассеянное рентгеновское излучение в них фиксируется на фотоплёнке или измеряется детекторами ядерных излучений (См. Детекторы ядерных излучений). В зависимости от состояния исследуемого образца и его свойств, а также от характера и объёма информации, которую необходимо получить, применяют различные методы Р. с. а. Монокристаллы, отбираемые для исследования атомной структуры, должны иметь размеры Рентгеновский структурный анализ 0,1 мм и по возможности обладать совершенной структурой. Исследованием дефектов в сравнительно крупных почти совершенных кристаллах занимается Рентгеновская топография, которую иногда относят к Р. с. а.
Метод Лауэ — простейший метод получения рентгенограмм от монокристаллов. Кристалл в эксперименте Лауэ неподвижен, а используемое рентгеновское излучение имеет непрерывный спектр. Расположение дифракционных пятен на лауэграммах (рис. 1) зависит от симметрии кристалла (См. Симметрия кристаллов) и его ориентации относительно падающего луча. Метод Лауэ позволяет установить принадлежность исследуемого кристалла к одной и 11 лауэвских групп симметрии и ориентировать его (т. е. определять направление кристаллографических осей) с точностью до нескольких угловых минут. По характеру пятен на лауэграммах и особенно появлению Астеризма можно выявить внутренние напряжения и некоторые др. дефекты кристаллической структуры. Методом Лауэ проверяют качество монокристаллов при выборе образца для его более полного структурного исследования.
Методы качания и вращения образца используют для определения периодов повторяемости (постоянной решётки) вдоль кристаллографического направления в монокристалле. Они позволяют, в частности, установить параметры а, b, с элементарной ячейки кристалла. В этом методе используют монохроматическое рентгеновское излучение, образец приводится в колебательное или вращательное движение вокруг оси, совпадающей с кристаллографическим направлением, вдоль которого и исследуют период повторяемости. Пятна на рентгенограммах качания и вращения, полученных в цилиндрических кассетах, располагаются на семействе параллельных линий. Расстояния между этими линиями, длина волны излучения и диаметр кассеты рентгеновской камеры позволяют вычислить искомый период повторяемости в кристалле. Условия Лауэ для дифракционных лучей в этом методе выполняются за счёт изменения углов, входящих в соотношения (1) при качании или вращении образца.
Рентгенгониометрические методы. Для полного исследования структуры монокристалла методами Р. с. а. необходимо не только установить положение, но и измерить интенсивности как можно большего числа дифракционных отражений, которые могут быть получены от кристалла при данной длине волны излучения и всех возможных ориентациях образца. Для этого дифракционную картину регистрируют на фотоплёнке в рентгеновском гониометре (См. Рентгеновский гониометр) и измеряют с помощью Микрофотометра степень почернения каждого пятна на рентгенограмме. В рентгеновском дифрактометре (См. Рентгеновский дифрактометр) можно непосредственно измерять интенсивность дифракционных отражений с помощью пропорциональных, сцинтилляционных и других счётчиков рентгеновских квантов. Чтобы иметь полный набор отражений, в рентгеновских гониометрах получают серию рентгенограмм. На каждой из них фиксируются дифракционные отражения, на миллеровские индексы которых накладывают определённые ограничения (например, на разных рентгенограммах регистрируются отражения типа hk0, hk1 и т.д.). Наиболее часто производят рентгеногониометрический эксперимент по методам Вайсенберга. Бюргера (рис. 2) и де Ионга — Боумена. Такую же информацию можно получить и с помощью рентгенограмм качания.
Для установления атомной структуры средней сложности (Рентгеновский структурный анализ 50—100 атомов в элементарной ячейке) необходимо измерить интенсивности нескольких сотен и даже тысяч дифракционных отражений. Эту весьма трудоёмкую и кропотливую работу выполняют автоматические микроденситометры и дифрактометры, управляемые ЭВМ, иногда в течение нескольких недель и даже месяцев (например, при анализе структур белков, когда число отражений возрастает до сотен тысяч). Применением в дифрактометре нескольких счётчиков, которые могут параллельно регистрировать отражения, время эксперимента удаётся значительно сократить. Дифрактометрические измерения превосходят фоторегистрацию по чувствительности и точности.
Метод исследования поликристаллов (Дебая - Шеррера метод). Металлы, сплавы, кристаллические порошки состоят из множества мелких монокристаллов данного вещества. Для их исследования используют монохроматическое излучение. Рентгенограмма (дебаеграмма) поликристаллов представляет собой несколько концентрических колец, в каждое из которых сливаются отражения от определённой системы плоскостей различно ориентированных монокристаллов. Дебаеграммы различных веществ имеют индивидуальный характер и широко используются для идентификации соединений (в том числе и в смесях). Р.с.а. поликристаллов позволяет определять фазовый состав образцов, устанавливать размеры и преимущественную ориентацию (текстурирование) зёрен в веществе, осуществлять контроль за напряжениями в образце и решать другие технические задачи.
Исследование аморфных материалов и частично упорядоченных объектов. Чёткую рентгенограмму с острыми дифракционными максимумами можно получить только при полной трёхмерной периодичности образца. Чем ниже степень упорядоченности атомного строения материала, тем более размытый, диффузный характер имеет рассеянное им рентгеновское излучение. Диаметр диффузного кольца на рентгенограмме аморфного вещества может служить для грубой оценки средних межатомных расстояний в нём. С ростом степени упорядоченности (см. Дальний порядок и ближний порядок) в строении объектов дифракционная картина усложняется и, следовательно, содержит больше структурной информации.
Метод малоуглового рассеяния позволяет изучать пространственные неоднородности вещества, размеры которых превышают межатомные расстояния, т.е. составляют от 5—10 Å до Рентгеновский структурный анализ 10 000 Å. Рассеянное рентгеновское излучение в этом случае концентрируется вблизи первичного пучка — в области малых углов рассеяния. Малоугловое рассеяние применяют для исследования пористых и мелкодисперсных материалов, сплавов и сложных биологических объектов: вирусов, клеточных мембран, хромосом. Для изолированных молекул белка и нуклеиновых кислот метод позволяет определить их форму, размеры, молекулярную массу; в вирусах — характер взаимной укладки составляющих их компонент: белка, нуклеиновых кислот, липидов; в синтетических полимерах — упаковку полимерных цепей; в порошках и сорбентах — распределение частиц и пор по размерам; в сплавах — возникновение и размеры фаз; в текстурах (в частности, в жидких кристаллах) — форму упаковки частиц (молекул) в различного рода надмолекулярные структуры. Рентгеновский малоугловой метод применяется и в промышленности при контроле процессов изготовления катализаторов, высокодисперсных углей и т.д. В зависимости от строения объекта измерения производят для углов рассеяния от долей минуты до нескольких градусов.
Определение атомной структуры по данным дифракции рентгеновских лучей. Расшифровка атомной структуры кристалла включает: установление размеров и формы его элементарной ячейки; определение принадлежности кристалла к одной из 230 федоровских (открытых Е. С. Федоровым (См. Фёдоров)) групп симметрии кристаллов (См. Симметрия кристаллов); получение координат базисных атомов структуры. Первую и частично вторую задачи можно решить методами Лауэ и качания или вращения кристалла. Окончательно установить группу симметрии и координаты базисных атомов сложных структур возможно только с помощью сложного анализа и трудоёмкой математической обработки значений интенсивностей всех дифракционных отражений от данного кристалла. Конечная цель такой обработки состоит в вычислении по экспериментальным данным значений электронной плотности ρ(х, у, z) в любой точке ячейки кристалла с координатами x, у, z. Периодичность строения кристалла позволяет записать электронную плотность в нём через Фурье ряд:

где V — объём элементарной ячейки, Fhkl — коэффициенты Фурье, которые в Р. с. а. называются структурными амплитудами, i =  hkl и связана с тем дифракционным отражением, которое определяется условиями (1). Назначение суммирования (2) — математически собрать дифракционные рентгеновские отражения, чтобы получить изображение атомной структуры. Производить таким образом синтез изображения в Р. с. а. приходится из-за отсутствия в природе линз для рентгеновского излучения (в оптике видимого света для этого служит собирающая линза).
hkl и связана с тем дифракционным отражением, которое определяется условиями (1). Назначение суммирования (2) — математически собрать дифракционные рентгеновские отражения, чтобы получить изображение атомной структуры. Производить таким образом синтез изображения в Р. с. а. приходится из-за отсутствия в природе линз для рентгеновского излучения (в оптике видимого света для этого служит собирающая линза).
hkl и связана с тем дифракционным отражением, которое определяется условиями (1). Назначение суммирования (2) — математически собрать дифракционные рентгеновские отражения, чтобы получить изображение атомной структуры. Производить таким образом синтез изображения в Р. с. а. приходится из-за отсутствия в природе линз для рентгеновского излучения (в оптике видимого света для этого служит собирающая линза). Дифракционное отражение — волновой процесс. Он характеризуется амплитудой, равной ∣Fhkl∣, и фазой αhkl (сдвигом фазы отражённой волны по отношению к падающей), через которую выражается структурная амплитуда: Fhkl =∣Fhkl ∣(cosαhkl + isinαhkl). Дифракционный эксперимент позволяет измерять только интенсивности отражений, пропорциональные ∣Fhkl∣2, но не их фазы. Определение фаз составляет основную проблему расшифровки структуры кристалла. Определение фаз структурных амплитуд в принципиальном отношении одинаково как для кристаллов, состоящих из атомов, так и для кристаллов, состоящих из молекул. Определив координаты атомов в молекулярном кристаллическом веществе, можно выделить составляющие его молекулы и установить их размер и форму.
Легко решается задача, обратная структурной расшифровке: вычисление по известной атомной структуре структурных амплитуд, а по ним — интенсивностей дифракционных отражений. Метод проб и ошибок, исторически первый метод расшифровки структур, состоит в сопоставлении экспериментально полученных ∣Fhkl∣эксп, с вычисленными на основе пробной модели значениями ∣Fhkl∣выч. В зависимости от величины фактора расходимости

пробная модель принимается или отвергается. В 30-х гг. были разработаны для кристаллических структур более формальные методы, но для некристаллических объектов метод проб и ошибок по-прежнему является практически единственным средством интерпретации дифракционной картины.
Принципиально новый путь к расшифровке атомных структур монокристаллов открыло применение т. н. функций Патерсона (функций межатомных векторов). Для построения функции Патерсона некоторой структуры, состоящей из N атомов, перенесём её параллельно самой себе так, чтобы в фиксированное начало координат попал сначала первый атом. Векторы от начала координат до всех атомов структуры (включая вектор нулевой длины до первого атома) укажут положение N максимумов функции межатомных векторов, совокупность которых называется изображением структуры в атоме 1. Добавим к ним ещё N максимумов, положение которых укажет N векторов от второго атома, помещенного при параллельном переносе структуры в то же начало координат. Проделав эту процедуру со всеми N атомами (рис. 3), мы получим N2 векторов. Функция, описывающая их положение, и есть функция Патерсона.
Для функции Патерсона Р (u, υ, ω) (u, υ, ω — координаты точек в пространстве межатомных векторов) можно получить выражение:

из которого следует, что она определяется модулями структурных амплитуд, не зависит от их фаз и, следовательно, может быть вычислена непосредственно по данным дифракционного эксперимента. Трудность интерпретации функции Р (u, υ, ω) состоит в необходимости нахождения координат N атомов из N2 её максимумов, многие из которых сливаются из-за перекрытий, возникающих при построении функции межатомных векторов. Наиболее прост для расшифровки Р (u, υ, ω) случай, когда в структуре содержится один тяжёлый атом и несколько лёгких. Изображение такой структуры в тяжёлом атоме будет значительно отличаться от др. её изображений. Среди различных методик, позволяющих определить модель исследуемой структуры по функции Патерсона, наиболее эффективными оказались так называемые суперпозиционные методы, которые позволили формализовать её анализ и выполнять его на ЭВМ.
Методы функции Патерсона сталкиваются с серьёзными трудностями при исследовании структур кристаллов, состоящих из одинаковых пли близких по атомному номеру атомов. В этом случае более эффективными оказались Так называемые прямые методы определения фаз структурных амплитуд. Учитывая тот факт, что значение электронной плотности в кристалле всегда положительно (или равно нулю), можно получить большое число неравенств, которым подчиняются коэффициенты Фурье (структурные амплитуды) функции ρ(x, у, z). Методами неравенств можно сравнительно просто анализировать структуры, содержащие до 20—40 атомов в элементарной ячейке кристалла. Для более сложных структур применяются методы, основанные на вероятностном подходе к проблеме: структурные амплитуды и их фазы рассматриваются как случайные величины; из физических представлений выводятся функции распределения этих случайных величин, которые дают возможность оценить с учётом экспериментальных значений модулей структурных амплитуд наиболее вероятные значения фаз. Эти методы также реализованы на ЭВМ и позволяют расшифровать структуры, содержащие 100—200 и более атомов в элементарной ячейке кристалла.
Итак, если фазы структурных амплитуд установлены, то по (2) может быть вычислено распределение электронной плотности в кристалле, максимумы этого распределения соответствуют положению атомов в структуре (рис. 4). Заключительное уточнение координат атомов проводится на ЭВМ Наименьших квадратов методом и в зависимости от качества эксперимента и сложности структуры позволяет получить их с точностью до тысячных долей Å (с помощью современного дифракционного эксперимента можно вычислять также количественные характеристики тепловых колебаний атомов в кристалле с учётом анизотропии этих колебаний). Р. с. а. даёт возможность установить и более тонкие характеристики атомных структур, например распределение валентных электронов в кристалле. Однако эта сложная задача решена пока только для простейших структур. Весьма перспективно для этой цели сочетание нейтронографических и рентгенографических исследований: нейтронографические данные о координатах ядер атомов сопоставляют с распределением в пространстве электронного облака, полученным с помощью Р. с. а. Для решения многих физических и химических задач совместно используют рентгеноструктурные исследования и резонансные методы.
Вершина достижений Р. с. а. — расшифровка трёхмерной структуры белков, нуклеиновых кислот и других макромолекул. Белки в естественных условиях, как правило, кристаллов не образуют. Чтобы добиться регулярного расположения белковых молекул, белки кристаллизуют и затем исследуют их структуру. Фазы структурных амплитуд белковых кристаллов можно определить только в результате совместных усилий рентгенографов и биохимиков. Для решения этой проблемы необходимо получить и исследовать кристаллы самого белка, а также его производных с включением тяжёлых атомов, причём координаты атомов во всех этих структурах должны совпадать.
О многочисленных применениях методов Р. с. а. для исследования различных нарушений структуры твёрдых тел под влиянием всевозможных воздействий см. в ст. Рентгенография материалов.
Лит.: Белов Н. В., Структурная кристаллография, М., 1951; Жданов Г. С., Основы рентгеноструктурного анализа, М. — Л., 1940; Джеймс Р., Оптические принципы дифракции рентгеновских лучей, пер. с англ., М., 1950; Бокий Г. Б., Порай-Кошиц М. А., Рентгеноструктурный анализ, М., 1964; Порай-Кошиц М. А., Практический курс рентгеноструктурного анализа, М., 1960: Китайгородский А. И., Теория структурного анализа, М., 1957; Липеон Г., Кокрен В., Определение структуры кристаллов, пер. с англ., М., 1961; Вайнштейн Б. К., Структурная электронография, М., 1956; Бэкон Дж., Дифракция нейтронов, пер. с англ., М., 1957; Бюргер М., Структура кристаллов и векторное пространство, пер. с англ., М., 1961; Гинье А., Рентгенография кристаллов, пер. с франц., М., 1961; Woolfson М. М., An introduction to X-ray crystallography, Camb., 1970: Ramachandran G. N., Srinivasan R., Fourier methode in crystallography, N. Y., 1970; Crystallographic computing, ed. F. R. Ahmed, Cph., 1970; Stout G. H., Jensen L. H., X-ray structure determination, N. Y. — L., [1968].
В. И. Симонов.
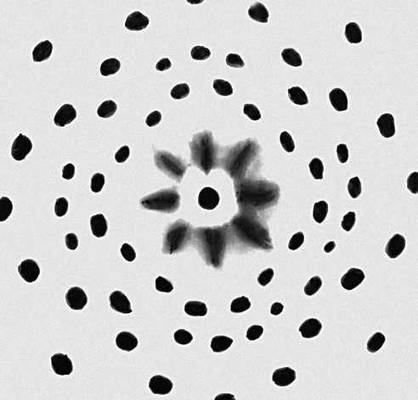
Рис. 1. Лауэграмма монокристалла NaCI. Каждое пятно представляет собой след рентгеновского дифракционного отражения. Диффузные радиальные пятна в центре вызваны рассеянием рентгеновских лучей на тепловых колебаниях кристаллической решётки.

Рис. 2. Рентгенограмма кристалла миоглобина.

Рис. 3. Схема построения функции Патерсона для структуры, состоящей из 3 атомов.

Рис. 9. а. Проекция на плоскость ab функции межатомных векторов минерала баотита [BA4Ti4 (Ti, Nb)4 [Si4O12] O16Cl]. Линии проведены через одинаковые интервалы значений функции межатомных векторов (линии равного уровня). б. Проекция электронной плотности баотита на плоскость ab, полученная расшифровкой функции межатомных векторов (a). Максимумы электронной плотности (сгущения линий равного уровня) отвечают положениям атомов в структуре. в. Изображение модели атомной структуры баотита. Каждый атом Si расположен внутри тетраэдра, образованного четырьмя атомами O; атомы Ti и Nb — в октаэдрах, составленных атомами O. Тетраэдры SiO4 и октаэдры Ti(Nb)O6 в структуре баотита соединены, как показано на рисунке. Часть элементарной ячейки кристалла, соответствующая рис. а и б, выделена штриховой линией. Точечные линии на рис. а и б определяют нулевые уровни значений соответствующих функций.
Большая советская энциклопедия. — М.: Советская энциклопедия. 1969—1978.