* * *
a progressive central neurodegenerative disorder; it may be inherited or sporadic, and inherited forms may show autosomal dominant or multifactorial inheritance. It is believed to be caused by defects in β-amyloid precursor protein metabolism, and is characterized by diffuse atrophy throughout the cerebral cortex with distinctive lesions called senile plaques and clumps of fibrils called neurofibrillary tangles. There is a loss of choline acetyltransferase activity in the cortex, and many of the degenerating neurons are cholinergic neurons of the hippocampus and other limbic areas. The plaques contain numerous proteins, including an altered form of Aβ amyloid and apolipoprotein E, and the tangles contain mainly hyperphosphorylated tau protein. The first signs of the disease are slight memory disturbance or changes in personality; deterioration progresses to profound dementia over 5 to 10 years on average. Women are affected twice as often as men, and onset may occur at any age; the disorder is currently divided into early-onset and late-onset forms, with the dividing age being approximately 65 years, but there is no clinical distinction between the two forms. Mutations associated with the early-onset autosomal dominant form have been identified in the β-amyloid precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2) genes, all of which lead to increased production of the altered form of Aβ amyloid. Both familial and sporadic late-onset forms have been associated with a particular allele (ε4) of the apolipoprotein E gene (APOE).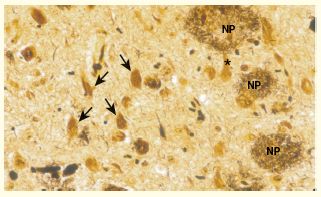
 Neurofibrillary tangles (arrows) and neuritic plaques (NP) in the neuronal cytoplasm in Alzheimer disease (silver stain).
Neurofibrillary tangles (arrows) and neuritic plaques (NP) in the neuronal cytoplasm in Alzheimer disease (silver stain).
Medical dictionary. 2011.