ADSORPTION
L’adsorption est le phénomène qui consiste en l’accumulation d’une substance à l’interface entre deux phases (gaz-solide, gaz-liquide, liquide-solide, liquide-liquide, solide-solide). Il a son origine dans les forces d’attraction intermoléculaires, de nature et d’intensité variées, qui sont responsables de la cohésion des phases condensées, liquides ou solides. Une molécule attirée inégalement par les autres molécules de deux phases trouvera une position énergétiquement favorable à la surface de la phase qui l’attire le plus; celle-ci sera appelée l’adsorbant , les molécules ainsi adsorbées constituant l’adsorbat . Si les conditions énergétiques ou cinétiques permettent à la molécule de pénétrer au sein de la phase adsorbante, il y a absorption .
Les cristaux constituent des édifices suffisamment rigides et stables pour que, le plus souvent, les molécules adsorbées ne modifient pas leur structure en surface: on s’intéressera alors surtout aux propriétés des molécules à l’état adsorbé. Par contre, la manifestation essentielle de l’adsorption à la surface des liquides est de modifier leur tension superficielle. Bien que les phénomènes fondamentaux soient les mêmes, l’adsorption se manifeste différemment sur les solides et sur les liquides; les méthodes d’étude et les domaines d’application sont différents et justifient des traitements distincts.
Adsorption gaz-solide
L’adsorption d’un gaz par un solide peut être mise en évidence expérimentalement d’autant plus facilement que l’aire de sa surface a été multipliée par son état de division. Si un solide cubique de 1 cm d’arête est divisé en petits cubes de 10 nm, l’aire de sa surface sera de 600 m2 et, si l’arête comprend une vingtaine de molécules, environ le quart des molécules du solide seront des molécules de surface. De tels états de division sont le plus souvent instables (ils sont thermodynamiquement métastables), mais on peut préparer facilement des charbons actifs dont l’aire spécifique peut être estimée à plus de 1 000 m2/g. L’aptitude du charbon de bois à retenir des gaz fut reconnue il y a plus de deux siècles.
Pour connaître la quantité adsorbée, le plus facile est de mesurer soit la quantité d’adsorbat qui disparaît de la phase gazeuse, soit l’accroissement de masse de l’adsorbant. Pour un couple adsorbant-adsorbat donné, cette quantité dépend de la température et de la pression, et on représente des isothermes d’adsorption en portant la quantité adsorbée à l’équilibre en fonction de la pression à température constante. D’autre part, la nature, l’état et la quantité des molécules adsorbées peuvent être étudiés actuellement par des méthodes analytiques très fines et très élaborées.
À partir d’isothermes d’adsorption à différentes températures, on peut déduire par une relation thermodynamique simple la variation d’énergie des molécules lors de leur passage de l’état gazeux à l’état adsorbé. On constate qu’en règle générale l’adsorption est exothermique (libérant la chaleur d’adsorption). D’après la règle de Le Châtelier, la quantité adsorbée à l’équilibre doit donc croître avec l’abaissement de la température ou avec l’augmentation de la pression. Dans certains cas, la quantité adsorbée est limitée et plus faible à basse température, lorsqu’on atteint les conditions de condensation massive de l’adsorbat, c’est-à-dire sa pression de vapeur.
La seule détermination des isothermes d’adsorption fournit quelques critères qui permettent, le plus souvent, de classer les adsorptions gaz-solide en deux catégories selon la nature des interactions qui retiennent l’adsorbat à la surface. S’il s’agit des forces physiques (forces de Van der Waals) responsables des attractions intermoléculaires dans tous les gaz et de leur condensation sous forme de liquides ou de cristaux moléculaires, on a affaire à une adsorption physique, ou physisorption . Si l’adsorbat se lie à la surface par des liaisons chimiques, il s’agira d’adsorption chimique, ou chimisorption .
Les chaleurs de physisorption sont comparables aux chaleurs de condensation des adsorbats, c’est-à-dire inférieures à environ 50 kJ/mole. L’adsorption est aisément réversible, comme le simple phénomène condensation-évaporation, et insensible à la nature chimique du couple adsorbant-adsorbat.
Les chaleurs de chimisorption sont comparables aux chaleurs de réactions chimiques, c’est-à-dire souvent supérieures à environ 100 kJ/mole. L’adsorption est conditionnée par les affinités chimiques entre adsorbant et adsorbat. Sa vitesse peut, comme pour les réactions chimiques, être très rapide ou très lente.
Physisorption
Un des buts poursuivis dans les recherches en physisorption depuis le début du siècle est d’imaginer des modèles capables de rendre compte de la forme des isothermes. Elles sont malheureusement extrêmement variées, tant par suite de la complexité des interactions que des multiples imperfections que présentent les surfaces réelles. C’est pourquoi, parmi les nombreuses équations proposées, aucune ne peut prétendre englober tous les types rencontrés expérimentalement. À un niveau élémentaire, on peut considérer qu’une molécule gazeuse entrant en collision avec la surface va y être retenue, c’est-à-dire adsorbée, grâce à leur attraction mutuelle, pendant un temps de séjour moyen 精 jusqu’à ce que les fluctuations thermiques lui fournissent l’énergie suffisante pour retourner dans la phase gazeuse. Le temps de séjour 精 dépend de la chaleur d’adsorption Q et de la température absolue T, selon la formule:

où R est la constante universelle des gaz et 精0 une constante de l’ordre de 10-13 s correspondant à la période d’oscillation de la molécule sur la surface. Le nombre de molécules adsorbées n a est égal au produit de la fréquence des collisions 益 des molécules gazeuses sur la surface par leur temps de séjour 精:

La théorie cinétique des gaz permet de calculer facilement 益, qui est proportionnel à la pression. La quantité adsorbée est donc simplement proportionnelle à la pression. Cette relation est vérifiée expérimentalement tant que les molécules adsorbées sont assez peu nombreuses pour n’occuper qu’une fraction faible de la surface. En effet, si les molécules qui viennent frapper une molécule déjà adsorbée rebondissent immédiatement en phase gazeuse (donc sans adsorption), seules celles qui arrivent sur la surface libre seront adsorbées. Ce modèle conduit à l’isotherme de Langmuir (1916): si on désigne par la fraction de surface occupée, définie par le rapport entre la quantité de gaz adsorbé V à la quantité Vm correspondant à une couche complète de molécules sur la surface, l’équation de l’isotherme s’écrit:

où P est la pression et K une constante dépendant du couple adsorbant-adsorbat et de la température. La courbe représentative est analogue à celle du type I de la figure 1. Elle est caractérisée par une concavité du côté de l’axe des pressions et montre que la quantité adsorbée croît asymptotiquement vers une valeur maximale correspondant à l’adsorption d’une seule couche de molécules (couche monomoléculaire, ou monocouche).
L’équation de Langmuir a l’avantage d’être d’une grande simplicité. Elle est cependant fréquemment inadaptée pour deux raisons essentielles. En premier lieu, les surfaces réelles sont hétérogènes, c’est-à-dire que tous les sites d’adsorption ne sont pas énergétiquement identiques. En second lieu, et surtout, la théorie néglige les attractions entre molécules adsorbées ainsi que l’attraction exercée par la surface au-delà de la première couche. Ces interactions peuvent conduire à la formation de plusieurs couches adsorbées (adsorption multimoléculaire). Elles se manifestent d’autant plus que l’adsorbat se trouve dans des conditions de température et de pression proches des conditions de sa condensation, sous forme de liquide ou de solide. On justifie ainsi une certaine normalisation des isothermes en portant en abscisse la pression relative P/P0, rapport entre la pression d’adsorption et la pression saturante de condensation de l’adsorbat. On a ainsi pu classer les isothermes en cinq types, dont les trois plus significatifs sont représentés schématiquement sur la figure 1. Les isothermes du type II sont, de loin, les plus fréquentes. Leur étude systématique pour de nombreux couples adsorbant-adsorbat a permis de conclure que la quantité adsorbée Vm au niveau de la courbure de l’isotherme en B (fig. 1) devait correspondre approximativement à l’adsorption d’une monocouche, l’adsorption multimoléculaire devenant prépondérante aux pressions plus élevées. La multiplication du nombre de molécules que représente Vm par l’aire occupée par une molécule (pouvant être estimée à partir de la masse volumique de l’adsorbat liquide ou solide) fournit directement l’aire de l’adsorbant. L’adsorption offre ainsi une méthode de mesure de l’aire de solides poreux ou pulvérulents. La difficulté d’attribuer à Vm une valeur bien définie, par suite de l’imprécision de localisation d’un point B, fut éludée par Brunauer, Emmett et Teller en 1938. Ils élaborèrent un modèle d’adsorption multimoléculaire (par une extension du modèle de Langmuir) conduisant à une équation d’isotherme (dite B.E.T., du nom de ses auteurs) qui permet de déterminer graphiquement une valeur de Vm . Cette équation n’a pas une importance fondamentale, car elle repose sur certaines hypothèses non fondées, voire incohérentes. Elle continue cependant d’être appliquée dans la mesure routinière des aires par adsorption.
Les isothermes du type III (fig. 1) sont assez rares (vapeur d’eau ou ammoniac sur graphite). Elles montrent une faible adsorption aux basses pressions, et une adsorption d’autant plus facile que la quantité déjà adsorbée est importante. Cela s’explique à la fois par une faible attraction adsorbant-adsorbat et par de fortes attractions entre molécules adsorbées. Dans un tel cas, la condensation de l’adsorbat est atteinte pour sa pression saturante alors que l’adsorption sur la surface est encore très limitée. Le phénomène de l’eau qui ne mouille pas le graphite est en relation avec cette particularité. Ce comportement est à opposer à celui qui correspond aux isothermes du type II, où le nombre de couches adsorbées tend apparemment vers l’infini quand on atteint la pression saturante (l’adsorbat mouille la surface).
Lorsque les adsorbants contiennent des pores de très petit diamètre (quelques nanomètres ou quelques dizaines de nanomètres), il peut se superposer à l’adsorption proprement dite une condensation capillaire de l’adsorbat. Elle se traduit souvent par des portions d’isothermes où les pressions d’équilibre apparent sont différentes lors de l’adsorption et de la désorption (c’est-à-dire lors de pressions respectivement croissantes et décroissantes). Ce phénomène, dit d’hystérèse, est très délicat à interpréter. On en tire cependant des renseignements sur la texture poreuse des adsorbants et des catalyseurs dont dépend la vitesse à laquelle les gaz peuvent y diffuser.
La plupart des adsorbants d’intérêt pratique ont non seulement une texture mal définie, mais une surface hétérogène (faces cristallines diverses et comportant quantité de défauts et d’imperfections). Ils sont donc mal désignés pour étudier l’adsorption dans des conditions bien définies. On obtient des résultats beaucoup plus significatifs avec des adsorbants à surface très homogène, tels que les solides bien cristallisés à structure lamellaire qui présentent une seule face cristalline développée préférentiellement (graphite, halogénures lamellaires...). À température suffisamment basse, l’isotherme d’adsorption de gaz simples se présente comme une succession de marches «verticales» et de hauteurs à peu près égales (fig. 2), traduisant la formation de couches distinctes superposées. L’adsorption débute aux basses pressions par une couche diluée de molécules mobiles sur la surface, analogue à un gaz à deux dimensions (2 D). À partir d’une certaine concentration superficielle, les interactions moléculaires provoquent l’apparition d’une phase condensée (2 D) qui occupe progressivement la surface à pression constante (transformation du premier ordre). Le même phénomène se reproduit pour les couches supérieures. La multiplicité des couches distinctes montre que l’énergie d’attraction du substrat se manifeste bien au-delà de la première couche (elle décroît en raison inverse de la puissance 3 de la distance). Leur nombre tend apparemment vers l’infini quand P atteint P0, le film adsorbé acquérant les mêmes propriétés énergétiques et structurales que l’adsorbat massif à trois dimensions (3 D). Mais il arrive que, lorsque P = P0, le nombre de couches condensées reste limité. Cela a lieu lorsque la face cristalline du substrat adsorbant impose au film condensé une structure n’ayant pas d’équivalent dans la phase 3 D de l’adsorbat, où les interactions moléculaires y sont le mieux satisfaites. Il n’y aura donc pas de nouvelle couche condensée avant que la phase 3 D n’apparaisse à P0 sous la forme de gouttelettes ou de petits cristaux.
L’étude détaillée de la formation de la première couche en fonction de la température a montré que la première marche peut se scinder en deux parties verticales, interprétables par des transitions gaz 2 Dliquide 2 D et liquide 2 Dsolide 2 D. On a pu aussi, comme pour les phases 3 D, mettre en évidence des points triples 2 D et des températures critiques 2 D; les phases solides 2 D peuvent aussi présenter des transitions entre différentes structures, en relation ou non avec celle du substrat. Ces phases 2 D sont surtout étudiées par des méthodes de diffraction d’électrons lents, de neutrons, de rayons X.
L’aspect lissé des isothermes d’adsorption habituelles s’explique par l’hétérogénéité des surfaces et par l’agitation thermique, aux températures où elles sont déterminées, qui estompe les différences énergétiques entre couches.
Chimisorption
En chimisorption, les liaisons dans les molécules sont profondément modifiées par suite de la formation de liaisons chimiques avec l’adsorbant. L’objectif des recherches concerne principalement la description des espèces adsorbées et l’étude de leur stabilité, en relation avec leur rôle essentiel d’intermédiaires dans les réactions catalysées par les solides.
La chimisorption traduit l’affinité chimique entre l’adsorbant et l’adsorbat. Par exemple, l’oxygène se chimisorbe sur les métaux et il y a une bonne corrélation entre ses chaleurs d’adsorption et les chaleurs de formation de leurs oxydes. La réaction s’arrête au stade superficiel lorsque des facteurs cinétiques ou thermodynamiques interdisent la formation d’un composé massif (3 D). Mais les atomes de surface ont des propriétés chimiques très particulières et sont capables de chimisorber des adsorbats avec lesquels ils ne peuvent former aucun composé connu: de nombreux métaux peuvent chimisorber des hydrocarbures ou autres molécules complexes. La formation des liaisons de chimisorption nécessite souvent la rupture de liaisons dans les molécules d’adsorbat, au point que celles-ci perdent leur identité: l’hydrogène et l’oxygène sont généralement adsorbés sur les métaux à l’état d’atomes, intensément liés à ceux du solide (la couche d’oxygène sur le tungstène est stable jusqu’à plus de 2 000 0C). Des molécules qui possèdent une liaison double peuvent se fixer par ouverture de celle-ci et formation de deux liaisons simples sur des atomes adjacents. La formation de ces liaisons entraîne des relations structurales étroites entre la couche adsorbée et l’adsorbant; les différentes faces cristallines présentent donc des comportements différents. Lorsque l’adsorbant et l’adsorbat ont fortement tendance à donner un composé (3 D) et que les atomes présentent une certaine mobilité (oxydes et sulfures métalliques), les atomes de la surface du substrat peuvent être déplacés perpendiculairement à celle-ci: on parle de «reconstruction» de la surface. Le composé de surface acquiert ainsi un début de structure tridimensionnelle apparentée à celle du composé massif. Ces aspects fondamentaux ne peuvent être étudiés que sur des faces de monocristaux et au moyen de techniques spécialisées. La nature et le mode de liaison des atomes adsorbés peuvent être déterminés à partir du spectre d’énergie des électrons qu’ils émettent sous l’effet d’excitations choisies, et les structures superficielles sont obtenues au moyen de la diffraction d’électrons lents.
La chimisorption ne pouvant, par nature, être multimoléculaire, et les interactions entre molécules adsorbées étant beaucoup plus faibles qu’avec le substrat, l’isotherme de Langmuir est en principe bien adaptée pour la représenter. Ce n’est cependant pas le cas pour les catalyseurs dont la surface est le plus souvent hétérogène. On a été ainsi amené à prendre en compte d’autres équations d’isothermes; mais celles-ci sont d’un usage trop complexe pour les calculs de cinétique et l’on se contente souvent d’utiliser l’équation de Langmuir, supposée applicable dans des domaines de température et de pression qui ne sont pas trop étendus.
Alors que la physisorption est aisément réversible à toutes températures, la chimisorption peut n’être possible que si, à l’instant de leur collision, une molécule d’adsorbat et un atome de surface possèdent, grâce à l’agitation thermique, un excès d’énergie appelé énergie d’activation. Cette énergie est nécessaire pour vaincre les contraintes momentanées qui précèdent l’état adsorbé final. La chimisorption n’atteint alors une vitesse appréciable qu’à une température suffisante, d’autant moins élevée que l’énergie d’activation est faible (voire nulle pour l’oxygène sur certains métaux).
Pour des adsorbants à surface hétérogène, la diminution de la chaleur d’adsorption en fonction du taux de recouvrement s’accompagne d’un accroissement de l’énergie d’activation. Cette conjonction des facteurs d’équilibre et des facteurs cinétiques conduit à des situations très difficiles à analyser.
Adsorption solutés-solides
La surface des solides est aussi capable d’adsorber des substances se trouvant en solution. Cette adsorption n’est pas fondamentalement différente de celle des gaz, la concentration du soluté jouant un rôle analogue à la pression, mais son traitement théorique est compliqué par l’adsorption compétitive du solvant. La couche physisorbée ne pourra être, le plus souvent, que monomoléculaire, car, au-delà de la première couche, les interactions soluté-solvant l’emportent sur les interactions soluté-surface.
Adsorption sur les liquides
La surface d’un liquide est, par nature, homogène; mais sa forme ou son étendue sont facilement modifiées sous l’action de contraintes. La résistance qu’elle oppose est une mesure de la tension superficielle du liquide. La présence d’un film adsorbé provoque une modification de cette tension superficielle, dont les manifestations pratiques sont extrêmement variées.
Les différents comportements d’une substance au contact de la surface d’un liquide présentent des analogies intéressantes avec les situations rencontrées sur les solides lorsque la substance est insoluble dans le liquide. Ce sont, par exemple, des composés organiques insolubles dans l’eau, de masse molaire assez grande et non volatils. On peut en déposer de très faibles quantités à la surface de l’eau à partir de solutions diluées dans un solvant volatil. On observe alors, suivant la nature de la goutte déposée, un des trois comportements typiques suivants:
– elle reste entièrement sous forme de lentille à la surface, sans étalement;
– elle s’étale sur toute la surface en un film mince, suffisamment épais pourtant pour montrer souvent des couleurs d’interférence;
– ou bien elle laisse une petite partie du liquide s’étendre sous forme d’une couche monomoléculaire dense, l’excès restant sous forme de lentille.
Le premier cas traduit une forte cohésion des molécules du liquide par rapport à leur interaction avec la surface de l’eau. Celle-ci peut, tout au plus, être recouverte par une couche diluée de molécules mobiles d’adsorbat, analogue à un gaz 2 D. La lentille est d’autant plus aplatie qu’elle est sollicitée radialement à sa périphérie par les molécules d’eau et que sa tension superficielle, qui tend à lui donner une forme sphérique, est moins forte. On observe cette évolution en passant des hydrocarbures lourds (huile de paraffine) à de plus légers (essence).
Cette situation est analogue à celle de l’adsorption d’un gaz sur un solide, lorsque, à la pression P0 en équilibre avec la phase 3 D de l’adsorbat, la couche adsorbée n’est encore constituée que d’un gaz 2 D.
Dans le deuxième cas, l’interaction entre la surface de l’eau et les molécules de la goutte l’emporte sur les forces de cohésion des molécules du liquide (cas de certains alcools par exemple), et la goutte s’étale complètement: les premières couches sont retenues à la surface par adsorption multimoléculaire; au-delà, c’est la gravité qui est responsable de l’uniformité d’épaisseur du film.
Le troisième cas est celui des molécules constituées d’une longue chaîne hydrocarbonée terminée par un groupement tel que 漣 COOH ou 漣 OH. Ces groupements «hydrophiles» sont caractérisés par de fortes interactions avec l’eau par liaison hydrogène, mais le caractère hydrophobe de la chaîne hydrocarbonée est suffisant pour que la substance soit insoluble. Les interactions avec la surface sont au mieux satisfaites par une orientation des molécules perpendiculairement à celle-ci, les extrémités hydrophiles étant en contact avec l’eau et les chaînes hydrocarbonées pointant vers l’extérieur. En présence d’un excès de substance déposé sous forme d’une goutte, il va donc se former à la surface de l’eau une couche monomoléculaire compacte de molécules ainsi orientées. Mais il n’y aura pas d’adsorption multimoléculaire, car les interactions adsorbant-adsorbat imposent au film adsorbé une structure telle que les interactions entre molécules d’adsorbat sont mieux satisfaites dans le liquide 3 D que dans une deuxième ou une troisième couche où le substrat n’attire presque plus. On retrouve donc un cas analogue à celui que l’on rencontre en physisorption sur les solides, lorsque le nombre de couches adsorbées à saturation est limité. Ces films qui ne sont pas mouillés par leur propre substance sont appelés «autophobes».
L’existence de films monomoléculaires à la surface de l’eau offre une possibilité d’étude particulière. En confinant le film derrière une barrière, mobile parallèlement à la surface de l’eau, on peut mesurer la force qu’il exerce sur celle-ci (balance de Langmuir). Cette force provient de la tendance du film à occuper la plus grande surface possible, ce qui se traduit par une pression superficielle (2 D). Si la quantité d’adsorbat déposé est suffisamment petite, le film aura les caractères d’un gaz 2 D: on constate effectivement en déplaçant la barrière que la pression superficielle est inversement proportionnelle à l’aire qu’il occupe. Une compression suffisante conduit à l’apparition d’une phase condensée orientée (2 D), qui croît ensuite latéralement à pression constante. Elle est éventuellement suivie d’autres transitions.
L’adsorption à la surface de l’eau se présente différemment lorsque l’adsorbat est dissous. Elle est accompagnée d’un abaissement de la tension superficielle de l’eau pour plusieurs raisons: les molécules d’eau subissent entre elles de très fortes interactions qui ne sont qu’à moitié satisfaites en surface; il faut dépenser beaucoup d’énergie pour amener une molécule de l’intérieur du liquide à la surface et accroître celle-ci; la tension superficielle tend à minimiser le nombre de molécules en surface. Il y a une compétition entre les molécules de soluté et de solvant pour ne pas être en surface. Lorsque les interactions entre molécules d’eau sont plus fortes qu’avec les molécules de soluté, celles-ci sont rejetées à la surface, avec diminution souvent très importante de la tension superficielle. Si, au contraire, l’eau présente de très fortes interactions avec le soluté (sels ionisés, sucres...), sa concentration est diminuée en surface et la tension superficielle n’est que très peu augmentée (ces solutés seront dits non tensio-actifs, par opposition aux précédents). La relation entre l’enrichissement (ou l’appauvrissement) 臨 en surface, la tension superficielle 靖 et la concentration c est donnée par une relation simple due à J. W. Gibbs (1878):
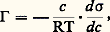
où R est la constante des gaz parfaits et T la température.
Applications
La physisorption sur les solides est fréquemment mise à profit pour la séparation et la purification des gaz ou la séparation de solutés dans les liquides. On utilise le plus souvent les charbons actifs, mais on dispose aussi d’adsorbants comportant des pores de dimensions moléculaires (comme les zéolites) qui leur confèrent une certaine sélectivité en fonction de la taille et de la forme des molécules. L’analyse chromatographique est basée sur les temps de séjour différents à l’état adsorbé des molécules d’un mélange gazeux ou liquide au cours de sa progression le long d’un adsorbant. La physisorption est enfin la seule méthode dont on dispose pratiquement pour mesurer l’aire des solides poreux ou pulvérulents (catalyseurs, pigments...).
La chimisorption est responsable de la présence constante de couches adsorbées (oxygène, eau, dioxyde de carbone...) à la surface des solides. Il en résulte de grandes difficultés expérimentales pour la préparation et l’étude des surfaces propres: sous une pression de 1 milliardième d’atmosphère, il peut suffire de quelques secondes pour que la surface soit contaminée par une couche chimisorbée; cela explique la nécessité de pressions des milliers de fois plus faibles (ultravide). Mais l’intérêt essentiel de la chimisorption est de permettre de mieux comprendre le rôle des intermédiaires réactionnels responsables de l’activité et de la spécificité des catalyseurs solides qui jouent un rôle considérable dans l’industrie chimique.
Dans les liquides, l’adsorption joue un rôle déterminant dans la stabilité des états de dispersion d’autres phases (mousses, émulsions, colloïdes). Ces propriétés sont mises à profit dans l’industrie des détergents.
L’adsorption joue un rôle important dans les phénomènes d’adhésion entre surfaces de solides. Elle intervient inévitablement dans le mécanisme de la croissance des cristaux, par migration de molécules à l’état adsorbé. Enfin, l’étude des domaines d’existence et de la structure des phases bidimensionnelles a approfondi nos connaissances sur les états de la matière.
adsorption [ atsɔrpsjɔ̃ ] n. f.
• 1904; lat. ad « sur » et rad. de absorption
♦ Phys. Rétention à la surface d'un solide (⇒ adsorbant) des molécules d'un gaz ou d'une substance en solution ou en suspension.
⊗ CONTR. Désorption.
● adsorption nom féminin (de adsorber) Phénomène par lequel des solides pulvérulents ou poreux, des solutions retiennent à leur surface des molécules, des ions en phase gazeuse ou liquide. ● adsorption (difficultés) nom féminin (de adsorber) Emploi Ne pas confondre ces deux mots, proches par le sens et par la prononciation, mais dont les domaines d'emploi sont bien distincts. 1. Absorption appartient à la langue courante : ivresse par absorption massive d'alcool. 2. Adsorption est un terme de la langue scientifique et technique : adsorption des gaz toxiques par le charbon de bois.
adsorption
n. f. PHYS, CHIM Fixation d'ions libres, d'atomes ou de molécules à la surface d'une substance. Ant. désorption.
⇒ADSORPTION, subst. fém.
CHIM., PHYS. Phénomène par lequel la surface d'un corps fixe, en les concentrant, les molécules libres ou dissoutes d'un liquide ou d'un gaz avec lesquels elle est en contact :
• 1. Les phénomènes d'adsorption. — On appelle ainsi le phénomène de la condensation de substances au niveau des surfaces de contact.
A. POLICARD, Précis d'histologie physiologique, 1922, p. 60.
• 2. Les électrolytes peuvent être absorbés par les colloïdes et entraîner la précipitation ou floculation de ceux-ci. Le précipité obtenu placé dans l'eau pure n'abandonne pas l'électrolyte qu'il recèle; l'absorption est irréversible, on lui donne le nom d'adsorption.
M. GASNIER, Dépôts métalliques directs et indirects, 1927, pp. 187-188.
• 3. Le raffinage utilise parfois les phénomènes d'adsorption pour éliminer des impuretés des produits pétrolifères.
J.-J. CHARTROU, Pétroles naturel et artificiels, 1931, p. 23.
• 4. Elle [la physico-chimie] en est encore à étudier par exemple les phénomènes d'adsorption qui sont susceptibles d'éclairer la fixation élective des aliments par le protoplasma.
PLANTEFOL, Cours de botanique et de biologie végétale, t. 1, 1931, p. 106.
• 5. L'adsorption :c'est une propriété physico-chimique fondamentale des solides et des liquides, utilisée en particulier pour la purification des corps et pour la catalyse. Crée une élévation de température.
Sc. 1962.
— Adsorption activée :
• 6. La « sorption » lente qui a souvent lieu aux températures élevées a été attribuée soit a l'« adsorption activée » soit à un phénomène de solution.
Journal de chimie et de physique, 1933, p. 654.
• 7. Un second type d'adsorption, appelé adsorption « activée » a lieu à des températures plus élevées,...
Journal de chimie et de physique, 1934, p. 100.
— ÉLECTR. ,,Risque de conduction électrique à la surface d'un isolant par suite de la condensation de l'humidité ambiante.`` (SIZ. 1968).
Prononc. ET ORTH. :[ ]. — Rem. Ne pas confondre avec absorption (cf. Ortho-vert 1966).
]. — Rem. Ne pas confondre avec absorption (cf. Ortho-vert 1966).
]. — Rem. Ne pas confondre avec absorption (cf. Ortho-vert 1966).Étymol. ET HIST. — 1922 phys. et chim., supra.
Dér. de adsorber; suff. -tion.
BBG. — BAULIG 1956. — BÉL. 1957. — FROMH.-KING 1968. — GARNIER-DEL. 1961 [1958]. — GRAND. 1962. — LAITIER 1969. — Pétrol. 1964. — PLAIS.-CAILL, 1958. — SIZ. 1968. — THOMAS 1956. — UV.-CHAPMAN 1956.
adsorption [atsɔʀpsjɔ̃] n. f.
❖
♦ Sc. Rétention à la surface d'un solide ou d'un liquide (dit adsorbant) des molécules d'un gaz ou d'une substance en solution ou en suspension.
0 Les autres (dilutions homéopathiques) sont de l'inconnu numéroté du fait des phénomènes d'adsorption (adhérence persistante des éléments dissous aux parois du flacon unique) et du mélange inévitable, en proportions inconnues, dans chaque dilution successive, de fractions des dilutions précédentes (…)
Pierre Vannier, l'Homéopathie, p. 121.
➪ tableau Vocabulaire de la chimie.
❖
CONTR. Désorption.
Encyclopédie Universelle. 2012.