- СПЕКТРАЛЬНЫЙ АНАЛИЗ
-
физич. методы качеств. .и количеств. определения состава в-ва, основанные на получении и исследовании его спектров. Основа С. а. — спектроскопия атомов и молекул, его классифицируют по целям анализа и типам спектров. Атомный С. а. (АСА) определяет элементный состав образца по атомным (ионным) спектрам испускания и поглощения; м о л е к у л я р н ы й С. а. (МСА) — мол. состав в-ва по мол. спектрам поглощения, люминесценции и комбинационного рассеяния света. Эмиссионный С. а. производят по спектрам испускания атомов, ионов и молекул, возбуждённым разл. источниками эл.-магн. излучения в . диапазоне от g-излучения до микроволнового. А б-сорбционный С. а. осуществляют по спектрам поглощения анализируемых объектов (атомов, молекул, ионов в-ва).Атомный С. а. (АСА)Качественный АСА осуществляют сопоставлением полученного спектра исследуемого в-ва со спектр. линиями элементов, приведёнными в спец. таблицах и атласах. В основе количественного АСА лежит соотношение, связывающее концентрацию с определяемого элемента с отношением интенсивностей линий определяемой примеси (I1) и линии сравнения (I2): I1/I2=асb (постоянные a и b определяются опытным путём), илиlg(I1/I2)=blgc+lga.С помощью стандартных образцов (не менее трёх) можно построить график зависимости lg(I1/I2) от Igc (градуировочный график, рис.) и определить по нему а и 6. Значения It и I2 можно получать непосредственно путём фотоэлектрич. измерений или путём фотометрирования (измерения плотности почернения) на микрофотометре линий определяемой примеси и линии сравнения при фоторегистрации.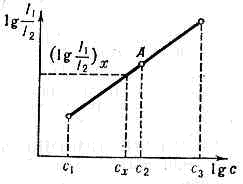 Градуировочный график (метод трёх эталонов).В эмиссионном АСА для получения спектров испускания исследуемого в-ва отбирают представит. пробу, отражающую его состав, и вводят её в источник излучения (атомизатор). Здесь тв. и жидкие пробы испаряются, соединение диссоциирует и свободные атомы (ионы) переходят в возбуждённое состояние. Испускаемое ими излучение раскладывается в спектр и регистрируется (или наблюдается визуально) с помощью спектрального прибора.Для возбуждения спектра в АСА используют разл. источники света и соответственно разл. способы введения в них образцов. Выбор источника зависит от конкретных условий анализа объекта. Тип источника и способ введения в него пробы составляют гл. содержание частных методик АСА. Первым искусств. источником света в АСА было пламя газовой горелки — источник. весьма удобный для быстрого и точного определения мн. элементов. Темп-ра пламён горючих газов невысока (от 2100К для смеси водород — воздух до 4500К для смеси кислород — циан). С помощью фотометрии пламенной определяют ок. 70 элементов по их аналитич. линиям, а также по мол. полосам соединений, образующихся в пламёнах.В эмиссионном АСА широко используются электрич. источники света. В электрич. дуге пост. тока между специально очищенными угольными электродами разл. формы, в каналы к-рых помещают исследуемое в-во в измельчённом состоянии, можно производить одновременно определение десятков элементов. Она обеспечивает относительно высокую темп-ру нагрева электродов и благоприятные условия возбуждения атомов пробы в дуговой плазме, однако точность этого метода невысока из-за нестабильности разряда. Повышая напряжение до 300—400 В или переходя к высоковольтной дуге (3—4 кВ), можно увеличить точность анализа.Более стабильные условия создаёт дуга перем. тока. В совр. генераторах дуги перем. тока можно получать разл. режимы возбуждения (низковольтную дугу, искру, ВЧ искру, дугу перем. тока, импульсный разряд и т. д.). Такие источники света с разл. режимами используют при определении металлов и трудно возбудимых элементов (углерод, галогены, газы, содержащиеся в металлах, и т. д.). Высоковольтная конденсиров. искра служит гл. обр. источником света при анализе металлов. Стабильность искрового разряда позволяет получать высокую воспроизводимость анализа, однако сложные процессы, происходящие на поверхностях электродов, приводят к изменению состава плазмы разряда. Чтобы устранить это явление, производят предварит. обжиг проб, нормируют форму и размеры проб и стандартных образцов.В эмиссионном АСА перспективно применение стабилизиров. форм электрич. разряда, получаемых в плазмотронах разл. конструкций, ВЧ индукционного разряда, СВЧ разряда, создаваемого магнетронными генераторами, ВЧ факельного разряда. С помощью разл. приемов введения анализируемых в-в в плазму этих разрядов (продувка порошков, распыление р-ров и т. д.) значительно повышена относит. точность анализа (до 0,5—3%), в т. ч. и компонентов сложных проб, содержание к-рых составляет десятки %. В нек-рых важных случаях анализа чистых в-в применение этих типов разряда снижает пределы определения примесей на 1—2 порядка (до 10-5—10-6 %).Для апализа чистых в-в, радиоактивных материалов, смесей газов, изотопного анализа, спектрально-изотопного определения газов в металлах и тв. телах и т. д. весьма перспективно оказалось использование разряда в полом катоде и безэлектродных ВЧ и СВЧ разрядов. В качестве источников возбуждения применяются также лазеры (см. ЛАЗЕРНАЯ СПЕКТРОСКОПИЯ).Атомно-абсорбционный С. а. (ААА) и атомно-флуоресцентный С. а. (АФА). В этих методах пробу также испаряют в атомизаторе (в пламени, графитовой трубке, плазме стабилизированного ВЧ и СВЧ разряда). В ААА свет от источника дискр. излучения, проходя через пар в-ва, ослабляется, и по степени ослабления интенсивностей линий определяемого элемента судят о концентрации его в пробе. ААА проводят на спец. спектрофотометрах; методика его проведения по сравнению с др. методами значительно проще, для него характерна высокая точность определения не только малых, но и больших концентраций элементов в пробах.В АФА ат. пары пробы облучают резонансным для исследуемого элемента излучением и регистрируют его флуоресценцию. Для нек-рых элементов (Zn, Cd, Hg и др.) относит, пределы обнаружения весьма малы (=10-5—10-6 %).АСА позволяет проводить измерение изотопного состава благодаря изотопному сдвигу спектр. линий (для большинства элементов требуются приборы высокой разрешающей способности, напр. эталон Фабри — Перо). Изотопный С. а. можно также проводить по электронно-колебательным спектрам молекул, определяя изотопные сдвиги полос, достигающие в некоторых случаях значительной величины.Экспрессные методы АСА широко применяются в пром-сти, с. х-ве, геологии и мн. др. областях нар. х-ва и науки. Значит. роль АСА играет в ат. технике, произ-ве чистых ПП материалов, сверхпроводников и т. д.К С. а. относится также анализ элементного состава в-ва по рентг. спектрам (см. СПЕКТРАЛЬНЫЙ АНАЛИЗ РЕНТГЕНОВСКИЙ), по спектрам оже- и фотоэлектронов ((см. ) Оже-спектроскопия и Фотоэлектронная спектроскопия), по спектрам фотопроводимости и др.Молекулярный спектральный анализ (МСА)В основе МСА лежит качеств. и количеств. сравнение измеренного спектра исследуемого образца со спектрами индивидуальных веществ. Соответственно различают качеств. и количеств. МСА. В МСА используют разл. виды молекулярных спектров: вращательные (микроволновая и длинноволновая ИК области спектра), колебательные и колебательно-вращательные (спектры поглощения и излучения в ср. ИК области, спектры комбинационного рассеяния света (КРС), спектры ИК флуоресценции), электронные, электронно-колебательные и электронно-колебательно-вращательные (спектры поглощения и пропускания в видимой и УФ областях, спектры флуоресценции). МСА позволяет проводить анализ малых количеств в-ва (до долей мкг и менее) в разл. агрегатных состояниях.Осн. факторы, определяющие возможности методов МСА:1) информативность метода. Условно выражается числом спектрально разрешаемых линий или полос в определ. интервале длин волн или частот исследуемого диапазона (для микроволн. диапазона оно =105, для ср. ИК области =103);2) кол-во измеренных спектров индивидуальных соединении;3) существование общих закономерностей между спектром в-ва и его мол. строением;4) чувствительность и избирательность метода;5) универсальность метода;6) простота и доступность измерений спектров.Качественный МСА устанавливает мол. состав исследуемого образца. Спектр молекулы явл. его однозначной хар-кой. Наиболее специфичны спектры в-в в газообразном состоянии с разрешённой вращат. структурой, к-рые исследуют с помощью спектр. приборов высокой разрешающей способности. Чаще всего используют спектры ИК поглощения и КРС в-в в жидком и тв. состояниях, а также спектры поглощения в видимой и УФ областях. Широкому внедрению метода КРС способствовало применение для их возбуждения лазерного излучения.Для повышения эффективности МСА в нек-рых случаях измерение спектров комбинируют с др. методами идентификации в-в. Так, всё большее распространение получает сочетание хроматографич. разделения в-в смесей с измерением ИК спектров поглощения выделенных компонентов.К качеств. МСА относится также т. н. структурный мол. анализ. Установлено, что молекулы, имеющие одинаковые структурные элементы, обнаруживают в спектрах поглощения и испускания (особенно колебательных) общие черты. Так, наличие сульфгидрильной группы (—SH) в структуре молекулы влечёт за собой появление в спектре полосы в интервале 2565—2575 см-1 нитрильной группы (—CN) — полосы 2200— 2300 см-1 и т. д. Присутствие этих характеристич. полос в колебат. спектрах в-в с общими структурными элементами объясняется характеристичностью частоты ((см. ) Характеристические частоты) и формы мн. мол. колебаний. Эта особенность колебательных (и в меньшей степени электронных) спектров позволяет определять структурный тип в-ва.Применение ЭВМ существенно упрощает и ускоряет качеств. анализ. В принципе его можно полностью автоматизировать, вводя показания спектр. приборов непосредственно в ЭВМ, в память к-рой заложены спектральные характеристич. признаки мн. в-в.Количественный МСА по спектрам поглощения основан на Бугера — Ламберта — Бера законе, устанавливающем связь между интенсивностями падающего I0 и прошедшего через в-во I света в зависимости от толщины поглощающего слоя l и концентрации в-ва с:I(l)=I0e-ccl.Коэфф, c явл. хар-кой поглощающей способности определяемого компонента для данной частоты излучения. Важное условие успешного проведения количеств. МСА — независимость c от с и постоянство c в измеряемом интервале частот, определяемом шириной щели спектрофотометра. МСА по спектрам поглощения проводят преим. для жидкостей и р-ров, для газов он значительно усложняется.В практич. МСА обычно измеряют т. н. оптич. плотность D:D = lnI0/I=ccl.Если смесь состоит из n в-в, не реагирующих друг с другом, то оптич. плотность смеси на частоте v аддитивна: D=Sni=1Div. Это позволяет проводить полный или частичный анализ многокомпонентных смесей. Задача в этом случае сводится к измерению значений оптич. плотности в m точках спектра смеси (m?n) и решения получаемой системы ур-ний:Dk=Sni=1Dki.Для количеств. МСА обычно пользуются спектрофотометрами, позволяющими производить измерения I(v) в сравнительно широком интервале v. Если полоса поглощения исследуемого в-ва достаточно изолирована и свободна от наложения полос др. компонентов смеси, исследуемый спектр. участок можно выделить, напр., при помощи интерференц. светофильтра. На его основе конструируют спец. анализаторы, используемые в промышленности.При количеств. MCA по спектрам КРС чаще всего интенсивность линий определяемого компонента смеси сравнивают с интенсивностью нек-рой линии стандартного в-ва, измеренной в тех же условиях (метод внеш. стандарта). В др. случаях стандартное в-во добавляют к исследуемому в определ. кол-ве (метод внутр. стандарта).Среди др. методов качеств. и количеств. МСА наибольшей чувствительностью обладает флуоресцентный анализ, однако он уступает методам колебат. спектроскопии в универсальности и избирательности. Количеств. МСА по спектрам флуоресценции основан на сравнении свечения р-ра исследуемого образца со свечением ряда эталонных р-ров близкой концентрации.Особое значение имеет флуоресцентный анализ с применением техники замороженных р-ров в спец. растворителях, напр. в парафинах (Шпольского эффект). Благодаря исключительно малой ширине спектр. линий в этом случае удаётся достичь высокой пороговой чувствительности обнаружения нек-рых многоатомных ароматич. соединений (= 10-11 г/см3).
Градуировочный график (метод трёх эталонов).В эмиссионном АСА для получения спектров испускания исследуемого в-ва отбирают представит. пробу, отражающую его состав, и вводят её в источник излучения (атомизатор). Здесь тв. и жидкие пробы испаряются, соединение диссоциирует и свободные атомы (ионы) переходят в возбуждённое состояние. Испускаемое ими излучение раскладывается в спектр и регистрируется (или наблюдается визуально) с помощью спектрального прибора.Для возбуждения спектра в АСА используют разл. источники света и соответственно разл. способы введения в них образцов. Выбор источника зависит от конкретных условий анализа объекта. Тип источника и способ введения в него пробы составляют гл. содержание частных методик АСА. Первым искусств. источником света в АСА было пламя газовой горелки — источник. весьма удобный для быстрого и точного определения мн. элементов. Темп-ра пламён горючих газов невысока (от 2100К для смеси водород — воздух до 4500К для смеси кислород — циан). С помощью фотометрии пламенной определяют ок. 70 элементов по их аналитич. линиям, а также по мол. полосам соединений, образующихся в пламёнах.В эмиссионном АСА широко используются электрич. источники света. В электрич. дуге пост. тока между специально очищенными угольными электродами разл. формы, в каналы к-рых помещают исследуемое в-во в измельчённом состоянии, можно производить одновременно определение десятков элементов. Она обеспечивает относительно высокую темп-ру нагрева электродов и благоприятные условия возбуждения атомов пробы в дуговой плазме, однако точность этого метода невысока из-за нестабильности разряда. Повышая напряжение до 300—400 В или переходя к высоковольтной дуге (3—4 кВ), можно увеличить точность анализа.Более стабильные условия создаёт дуга перем. тока. В совр. генераторах дуги перем. тока можно получать разл. режимы возбуждения (низковольтную дугу, искру, ВЧ искру, дугу перем. тока, импульсный разряд и т. д.). Такие источники света с разл. режимами используют при определении металлов и трудно возбудимых элементов (углерод, галогены, газы, содержащиеся в металлах, и т. д.). Высоковольтная конденсиров. искра служит гл. обр. источником света при анализе металлов. Стабильность искрового разряда позволяет получать высокую воспроизводимость анализа, однако сложные процессы, происходящие на поверхностях электродов, приводят к изменению состава плазмы разряда. Чтобы устранить это явление, производят предварит. обжиг проб, нормируют форму и размеры проб и стандартных образцов.В эмиссионном АСА перспективно применение стабилизиров. форм электрич. разряда, получаемых в плазмотронах разл. конструкций, ВЧ индукционного разряда, СВЧ разряда, создаваемого магнетронными генераторами, ВЧ факельного разряда. С помощью разл. приемов введения анализируемых в-в в плазму этих разрядов (продувка порошков, распыление р-ров и т. д.) значительно повышена относит. точность анализа (до 0,5—3%), в т. ч. и компонентов сложных проб, содержание к-рых составляет десятки %. В нек-рых важных случаях анализа чистых в-в применение этих типов разряда снижает пределы определения примесей на 1—2 порядка (до 10-5—10-6 %).Для апализа чистых в-в, радиоактивных материалов, смесей газов, изотопного анализа, спектрально-изотопного определения газов в металлах и тв. телах и т. д. весьма перспективно оказалось использование разряда в полом катоде и безэлектродных ВЧ и СВЧ разрядов. В качестве источников возбуждения применяются также лазеры (см. ЛАЗЕРНАЯ СПЕКТРОСКОПИЯ).Атомно-абсорбционный С. а. (ААА) и атомно-флуоресцентный С. а. (АФА). В этих методах пробу также испаряют в атомизаторе (в пламени, графитовой трубке, плазме стабилизированного ВЧ и СВЧ разряда). В ААА свет от источника дискр. излучения, проходя через пар в-ва, ослабляется, и по степени ослабления интенсивностей линий определяемого элемента судят о концентрации его в пробе. ААА проводят на спец. спектрофотометрах; методика его проведения по сравнению с др. методами значительно проще, для него характерна высокая точность определения не только малых, но и больших концентраций элементов в пробах.В АФА ат. пары пробы облучают резонансным для исследуемого элемента излучением и регистрируют его флуоресценцию. Для нек-рых элементов (Zn, Cd, Hg и др.) относит, пределы обнаружения весьма малы (=10-5—10-6 %).АСА позволяет проводить измерение изотопного состава благодаря изотопному сдвигу спектр. линий (для большинства элементов требуются приборы высокой разрешающей способности, напр. эталон Фабри — Перо). Изотопный С. а. можно также проводить по электронно-колебательным спектрам молекул, определяя изотопные сдвиги полос, достигающие в некоторых случаях значительной величины.Экспрессные методы АСА широко применяются в пром-сти, с. х-ве, геологии и мн. др. областях нар. х-ва и науки. Значит. роль АСА играет в ат. технике, произ-ве чистых ПП материалов, сверхпроводников и т. д.К С. а. относится также анализ элементного состава в-ва по рентг. спектрам (см. СПЕКТРАЛЬНЫЙ АНАЛИЗ РЕНТГЕНОВСКИЙ), по спектрам оже- и фотоэлектронов ((см. ) Оже-спектроскопия и Фотоэлектронная спектроскопия), по спектрам фотопроводимости и др.Молекулярный спектральный анализ (МСА)В основе МСА лежит качеств. и количеств. сравнение измеренного спектра исследуемого образца со спектрами индивидуальных веществ. Соответственно различают качеств. и количеств. МСА. В МСА используют разл. виды молекулярных спектров: вращательные (микроволновая и длинноволновая ИК области спектра), колебательные и колебательно-вращательные (спектры поглощения и излучения в ср. ИК области, спектры комбинационного рассеяния света (КРС), спектры ИК флуоресценции), электронные, электронно-колебательные и электронно-колебательно-вращательные (спектры поглощения и пропускания в видимой и УФ областях, спектры флуоресценции). МСА позволяет проводить анализ малых количеств в-ва (до долей мкг и менее) в разл. агрегатных состояниях.Осн. факторы, определяющие возможности методов МСА:1) информативность метода. Условно выражается числом спектрально разрешаемых линий или полос в определ. интервале длин волн или частот исследуемого диапазона (для микроволн. диапазона оно =105, для ср. ИК области =103);2) кол-во измеренных спектров индивидуальных соединении;3) существование общих закономерностей между спектром в-ва и его мол. строением;4) чувствительность и избирательность метода;5) универсальность метода;6) простота и доступность измерений спектров.Качественный МСА устанавливает мол. состав исследуемого образца. Спектр молекулы явл. его однозначной хар-кой. Наиболее специфичны спектры в-в в газообразном состоянии с разрешённой вращат. структурой, к-рые исследуют с помощью спектр. приборов высокой разрешающей способности. Чаще всего используют спектры ИК поглощения и КРС в-в в жидком и тв. состояниях, а также спектры поглощения в видимой и УФ областях. Широкому внедрению метода КРС способствовало применение для их возбуждения лазерного излучения.Для повышения эффективности МСА в нек-рых случаях измерение спектров комбинируют с др. методами идентификации в-в. Так, всё большее распространение получает сочетание хроматографич. разделения в-в смесей с измерением ИК спектров поглощения выделенных компонентов.К качеств. МСА относится также т. н. структурный мол. анализ. Установлено, что молекулы, имеющие одинаковые структурные элементы, обнаруживают в спектрах поглощения и испускания (особенно колебательных) общие черты. Так, наличие сульфгидрильной группы (—SH) в структуре молекулы влечёт за собой появление в спектре полосы в интервале 2565—2575 см-1 нитрильной группы (—CN) — полосы 2200— 2300 см-1 и т. д. Присутствие этих характеристич. полос в колебат. спектрах в-в с общими структурными элементами объясняется характеристичностью частоты ((см. ) Характеристические частоты) и формы мн. мол. колебаний. Эта особенность колебательных (и в меньшей степени электронных) спектров позволяет определять структурный тип в-ва.Применение ЭВМ существенно упрощает и ускоряет качеств. анализ. В принципе его можно полностью автоматизировать, вводя показания спектр. приборов непосредственно в ЭВМ, в память к-рой заложены спектральные характеристич. признаки мн. в-в.Количественный МСА по спектрам поглощения основан на Бугера — Ламберта — Бера законе, устанавливающем связь между интенсивностями падающего I0 и прошедшего через в-во I света в зависимости от толщины поглощающего слоя l и концентрации в-ва с:I(l)=I0e-ccl.Коэфф, c явл. хар-кой поглощающей способности определяемого компонента для данной частоты излучения. Важное условие успешного проведения количеств. МСА — независимость c от с и постоянство c в измеряемом интервале частот, определяемом шириной щели спектрофотометра. МСА по спектрам поглощения проводят преим. для жидкостей и р-ров, для газов он значительно усложняется.В практич. МСА обычно измеряют т. н. оптич. плотность D:D = lnI0/I=ccl.Если смесь состоит из n в-в, не реагирующих друг с другом, то оптич. плотность смеси на частоте v аддитивна: D=Sni=1Div. Это позволяет проводить полный или частичный анализ многокомпонентных смесей. Задача в этом случае сводится к измерению значений оптич. плотности в m точках спектра смеси (m?n) и решения получаемой системы ур-ний:Dk=Sni=1Dki.Для количеств. МСА обычно пользуются спектрофотометрами, позволяющими производить измерения I(v) в сравнительно широком интервале v. Если полоса поглощения исследуемого в-ва достаточно изолирована и свободна от наложения полос др. компонентов смеси, исследуемый спектр. участок можно выделить, напр., при помощи интерференц. светофильтра. На его основе конструируют спец. анализаторы, используемые в промышленности.При количеств. MCA по спектрам КРС чаще всего интенсивность линий определяемого компонента смеси сравнивают с интенсивностью нек-рой линии стандартного в-ва, измеренной в тех же условиях (метод внеш. стандарта). В др. случаях стандартное в-во добавляют к исследуемому в определ. кол-ве (метод внутр. стандарта).Среди др. методов качеств. и количеств. МСА наибольшей чувствительностью обладает флуоресцентный анализ, однако он уступает методам колебат. спектроскопии в универсальности и избирательности. Количеств. МСА по спектрам флуоресценции основан на сравнении свечения р-ра исследуемого образца со свечением ряда эталонных р-ров близкой концентрации.Особое значение имеет флуоресцентный анализ с применением техники замороженных р-ров в спец. растворителях, напр. в парафинах (Шпольского эффект). Благодаря исключительно малой ширине спектр. линий в этом случае удаётся достичь высокой пороговой чувствительности обнаружения нек-рых многоатомных ароматич. соединений (= 10-11 г/см3).
Физический энциклопедический словарь. — М.: Советская энциклопедия. Главный редактор А. М. Прохоров. 1983.
- СПЕКТРАЛЬНЫЙ АНАЛИЗ
-
- совокупность методов определения элементногои молекулярного состава и строения веществ по их спектрам. С помощью С. а. определяют как осн. компоненты, составляющие 50- 60% вещества анализируемыхобъектов, так и незначит. примеси в них (до
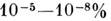 и менее). С. а. - наиб. распространённый аналитич. метод, св. 20- 30% всеханализов выполняется с помощью этого метода, в т. ч. контроль состава сплавовв металлургии, автомоб. и авиац. пром-сти, технологии переработки руд, анализ экологич. объектов и материалов высокой чистоты, хим., биол. и мед. исследования. Особо важное значение С. а. имеет при поисках полезных ископаемых.
и менее). С. а. - наиб. распространённый аналитич. метод, св. 20- 30% всеханализов выполняется с помощью этого метода, в т. ч. контроль состава сплавовв металлургии, автомоб. и авиац. пром-сти, технологии переработки руд, анализ экологич. объектов и материалов высокой чистоты, хим., биол. и мед. исследования. Особо важное значение С. а. имеет при поисках полезных ископаемых.Основа С. а.- спектроскопия атомов и молекул; его классифицируютпо целям анализа и типам спектров. В атомном С. а. (АСА) определяют элементныйсостав образцов по атомным (ионным) спектрам испускания и поглощения; вмолекулярном С. а. (МСА) - молекулярный состав вещества по молекулярнымспектрам поглощения, испускания, отражения, люминесценции и комбинационногорассеяния света. Эмиссионный С. а. проводят по спектрам испусканиявозбуждённых атомов, ионов и молекул. Абсорбционный С. а. осуществляютпо спектрам поглощения анализируемых объектов. В С. а. часто сочетают неск. спектральных методов, а также применяют др. аналитич. методы, что расширяетвозможности анализа. Для получения спектров используют разл. типы спектральныхприборов в зависимости от целей и условий анализа. Обработка эксперим. данных может производиться на ЭВМ, встроенных в спектральный прибор.
Атомный спектральный анализ Различают два осн. варианта атомногоС. а.- атомно-эмиссионный (АЭСА) и атомно-абсорбционный (ААА).
Атомно-эмиссионный спектральный анализ основан на зависимости 1 =f(с) интенсивности 1 спектральной линии испускания (эмиссии)определяемого элемента х от его концентрации в анализируемом объекте:
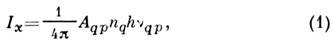
где
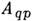 -вероятность квантового перехода из состояния q в состояние р,nq - концентрация атомов, находящихся в состоянии q висточнике излучения (исследуемом веществе),
-вероятность квантового перехода из состояния q в состояние р,nq - концентрация атомов, находящихся в состоянии q висточнике излучения (исследуемом веществе),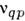 - частота квантового перехода. Если в зоне излучения выполняется локальноетермодинамическое равновесие, концентрация электронов п e14-1015 и их распределение по скоростям максвелловское, то
- частота квантового перехода. Если в зоне излучения выполняется локальноетермодинамическое равновесие, концентрация электронов п e14-1015 и их распределение по скоростям максвелловское, то 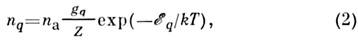
где n а - концентрация невозбуждённых атомов определяемогоэлемента в области излучения, gq - статистический вес состояния q,Z - статистическая сумма по состояниям q, причём
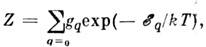
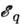 - энергиявозбуждения уровня q. Т. о., искомая концентрация n а- ф-ция темп-ры, к-рая практически не может строго контролироваться. Поэтомуобычно измеряют интенсивность аналитич. линии относительно нек-рого внутр. стандарта, присутствующего в анализируемом объекте в известной концентрацииn ст. Если стандартная линия близка к аналитической, то
- энергиявозбуждения уровня q. Т. о., искомая концентрация n а- ф-ция темп-ры, к-рая практически не может строго контролироваться. Поэтомуобычно измеряют интенсивность аналитич. линии относительно нек-рого внутр. стандарта, присутствующего в анализируемом объекте в известной концентрацииn ст. Если стандартная линия близка к аналитической, то 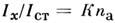 (K - постоянная величина). Эта зависимость используется в С. а. в тех случаях, когда отсутствует самообращение используемых линий.
(K - постоянная величина). Эта зависимость используется в С. а. в тех случаях, когда отсутствует самообращение используемых линий.В АЭСА применяются в осн. спектральные приборы с фоторегистрацией(спектрографы) и фотоэлектрич. регистрацией (квантометры). Излучение исследуемогообразца направляется на входную щель прибора с помощью системы линз, попадаетна диспергирующее устройство (призма или дифракц. решётка) и после монохроматизациифокусируется системой линз в фокальной плоскости, где располагается фотопластинкаили система выходных щелей (квантометр), за к-рыми установлены фотоэлементыили фотоумножители. При фоторегистрации интенсивности линий определяютпо плотности почернения S, измеряемой микрофотометром:
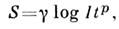
где р - т. н. константа Шварцшильда,
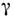 - фактор контрастности; t - время экспозиции.
- фактор контрастности; t - время экспозиции.В АЭСА исследуемое вещество должно находиться в состоянии атомного газа. Обычно атомизация и возбуждение атомов осуществляются одновременно - висточниках света. Для анализа металлов, сплавов и др. проводников чащевсего используют дуговой разряд или искровой разряд, гдев качестве электродов служат сами анализируемые пробы. Дуговой разряд применяетсяи для анализа непроводящих веществ. В этом случае порошкообразную пробупомещают в углубление в графитовом электроде (метод испарения) или с помощьюразл. устройств вводят порошок в плазму дугового разряда между горизонтальнорасположенными графитовыми электродами. Применяется также введение порошкообразныхпроб в дуговые плазмотроны.
При АЭСА растворов в качестве источников возбуждающего света применяютпламя горючих газов (смеси ацетилен - кислород, ацетилен - закись азотаи др.). В качестве источников света начали использовать также безэлектродныйразряд и особенно индуктивносвязанную плазму. Во всех случаях растворв виде аэрозоля потоком аргона вводят в зону возбуждения спектра (темп-ра2500-3000 К в пламенах и 6000- 10000 К в плазме разряда), где происходитвысушивание, испарение и атомизация аэрозоля.
Процесс атомизации в методах АЭСА обычно носит термич. характер, чтопозволяет сделать нек-рые обобщения. В реальных условиях, учитывающих кинетикупроцесса, для частиц, находящихся в зоне с темп-рой ТT кип(T кип - темп-pa кипения), зависимость кол-ва испарившихсячастиц от времени описывается ур-нием:
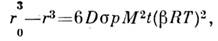
где r - радиус частицы, D - коэф. диффузии,
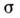 -поверхностное натяжение раствора, р- давление насыщенных паров, М- мол. масса,
-поверхностное натяжение раствора, р- давление насыщенных паров, М- мол. масса,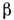 - плотность. Пользуясь этим ур-нием, можно найти кол-во вещества, испарившеесяза время t.
- плотность. Пользуясь этим ур-нием, можно найти кол-во вещества, испарившеесяза время t.Если при этом молекула состоит из элементов п 1 и n2,то степень атомизации может быть рассчитана по ур-нию:
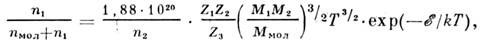
где М 1 и M2 - ат. массы элементов п 1 и n2; Z1 и Z2 - статистич. суммы по состояниям этих элементов, M МОЛ - мол. массаатомизирующейся молекулы, Z3 - статистич. сумма по еёсостояниям,
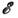 -энергия диссоциации молекулы. Такого типа расчёты позволяют найти концентрациюатомов определяемого элемента п а в ур-нии (2) и определитьеё связь с интенсивностью аналитич. линии. Необходимость учитывать взаимодействиеопределяемого элемента с окружающей средой, др. компонентами анализируемоговещества, ионизацию атомов определяемого элемента и др. эффекты значительноусложняет картину испарения и атомизации исследуемого вещества. С цельюоблегчения С. а. создаются спец. программы расчёта на ЭВМ достаточно сложныхреакций в газовой и конденсированных фазах при заданных темп-ре и давлении.
-энергия диссоциации молекулы. Такого типа расчёты позволяют найти концентрациюатомов определяемого элемента п а в ур-нии (2) и определитьеё связь с интенсивностью аналитич. линии. Необходимость учитывать взаимодействиеопределяемого элемента с окружающей средой, др. компонентами анализируемоговещества, ионизацию атомов определяемого элемента и др. эффекты значительноусложняет картину испарения и атомизации исследуемого вещества. С цельюоблегчения С. а. создаются спец. программы расчёта на ЭВМ достаточно сложныхреакций в газовой и конденсированных фазах при заданных темп-ре и давлении.В источниках излучения чаще всего не соблюдается термодинамич. равновесие, поэтому эти расчёты могут использоваться лишь при выборе оптим. условийанализа. В АЭСА применяют эмпирич. метод, заключающийся в эксперим. построениианалитич. ф-ции
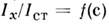 с помощью серии стандартных образцов анализируемого материала с заранееточно известными содержаниями определяемого элемента. Такие образцы либоизготовляют специально, либо заранее в неск. образцах устанавливают концентрациюэтого элемента точными методами. Измеряя затем аналитич. сигнал
с помощью серии стандартных образцов анализируемого материала с заранееточно известными содержаниями определяемого элемента. Такие образцы либоизготовляют специально, либо заранее в неск. образцах устанавливают концентрациюэтого элемента точными методами. Измеряя затем аналитич. сигнал 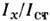 , находят содержание определяемого элемента в пробе.
, находят содержание определяемого элемента в пробе.Структура и физ.-хим. свойства анализируемого и стандартного объектовмогут оказаться неадекватными (различны, напр., условия парообразованиястепени атомизации, условий возбуждения). Эти различия приходится учитыватьпри С. а. В таких случаях используют метод факторного статистич. планированияэксперимента. В результате экспериментов получают т. н. ур-ния регрессии, учитывающие влияние на интенсивность аналитич. линий концентраций всехэлементов, составляющих пробу, и устанавливают концентрацию анализируемогоэлемента с помощью этих ур-ний. Совр. многоканальные квантометры позволяютодновременно измерять интенсивность большого числа спектральных линий. На основе этих эксперим. данных с помощью ЭВМ можно решать довольно сложныеслучаи анализа, однако за счёт измерения неск. линий случайная погрешностьопределения С. возрастает.
Атомно-абсорбционный анализ (ААА) основан на зависимости аналитич. сигнала(абсорбционности)
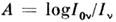 (где
(где 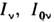 - интенсивности падающего и прошедшего сквозь образец света) от концентрации( Бугера- Ламберта - Берa закон):
- интенсивности падающего и прошедшего сквозь образец света) от концентрации( Бугера- Ламберта - Берa закон):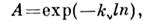
где kv - коэф. поглощения на частоте v, l - эфф. длина светового пути в области поглощения, п - концентрация атомованализируемого элемента в парах.
Схема установки ААА включает: независимый источник излучения света счастотой v, равной частоте аналитич. линии определяемого элемента; атомизатор, преобразующий пробу в атомарный пар; спектрофотометр. Свет, прошедший сквозьатомный пар, системой линз направляется на входную щель спектрофотометра, интенсивность аналитич. спектральной линии
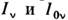 на выходе регистрируется фотоэлектрич. методом. Поскольку естественнаяширина спектральной линии, постоянна, зависит только от времени жизнивозбуждённого состояния и обычно пренебрежимо мала, разница контуров линиииспускания и поглощения определяется в осн. допплеровским
на выходе регистрируется фотоэлектрич. методом. Поскольку естественнаяширина спектральной линии, постоянна, зависит только от времени жизнивозбуждённого состояния и обычно пренебрежимо мала, разница контуров линиииспускания и поглощения определяется в осн. допплеровским 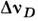 и лоренцевским
и лоренцевским 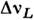 уширениями:
уширениями: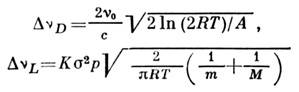
(здесь р - давление, с - скорость света, т - атомная, М- молекулярная массы,
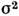 - эфф. сечение столкновений, приводящих к уширению, К - константа).Т. о., ширины контуров линий поглощения и испускания могут быть различнымив зависимости от давления, темп-ры и состава газовой фазы в источнике излученияи в поглощающей ячейке, что отразится на виде ф-ции
- эфф. сечение столкновений, приводящих к уширению, К - константа).Т. о., ширины контуров линий поглощения и испускания могут быть различнымив зависимости от давления, темп-ры и состава газовой фазы в источнике излученияи в поглощающей ячейке, что отразится на виде ф-ции 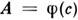 и может привести к неоднозначности результатов С. а. До нек-рой степениэто удаётся устранить достаточно сложными приёмами. В методе Уолша применяютлампы с полым катодом (ЛПК), к-рые излучают спектральные линии значительноболее узкие, чем линии поглощения атомов определяемых элементов в обычныхпоглощающих ячейках. В результате зависимость
и может привести к неоднозначности результатов С. а. До нек-рой степениэто удаётся устранить достаточно сложными приёмами. В методе Уолша применяютлампы с полым катодом (ЛПК), к-рые излучают спектральные линии значительноболее узкие, чем линии поглощения атомов определяемых элементов в обычныхпоглощающих ячейках. В результате зависимость 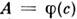 в довольно широких пределах значений А (0 -0,3) оказывается простойлинейной ф-цией.
в довольно широких пределах значений А (0 -0,3) оказывается простойлинейной ф-цией.В качестве атомизатора в ААА используют разл. пламена на основе смесейводород - кислород, ацетилен - воздух, ацетилен - закись азота и др. Анализуподвергают аэрозоль раствора пробы, вдуваемый в горящее пламя. Последовательноизмеряют интенсивности
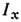 и I0 света, прошедшего сквозь пламя во время подачи аэрозоляи без его подачи. В совр. приборах измерение
и I0 света, прошедшего сквозь пламя во время подачи аэрозоляи без его подачи. В совр. приборах измерение 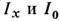 автоматизировано. В нек-рых случаях процессы испарения и последующей атомизациипробы из-за низкой темп-ры пламён ( Т ~3000 К) в газовой фазе происходятне полностью. Процессы испарения частиц аэрозоля и степень атомизации впламени сильно зависят также от состава пламени (соотношения горючего иокислителя), а также от состава раствора аэрозоля. Хорошую воспроизводимостьаналитич. сигнала (в лучших случаях Sr составляет 0,01-0,02)удаётся получать, применяя в качестве источников ЛПК, излучение к-рогообладает высокой стабильностью, и осуществляя процессы испарения и атомизациив пламени.
автоматизировано. В нек-рых случаях процессы испарения и последующей атомизациипробы из-за низкой темп-ры пламён ( Т ~3000 К) в газовой фазе происходятне полностью. Процессы испарения частиц аэрозоля и степень атомизации впламени сильно зависят также от состава пламени (соотношения горючего иокислителя), а также от состава раствора аэрозоля. Хорошую воспроизводимостьаналитич. сигнала (в лучших случаях Sr составляет 0,01-0,02)удаётся получать, применяя в качестве источников ЛПК, излучение к-рогообладает высокой стабильностью, и осуществляя процессы испарения и атомизациив пламени.В ААА (как и в АЭСА) эмпирически строят зависимость
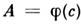 с помощью образцов, содержащих точно известные кол-ва определяемого элемента. Если общий состав этих образцов идентичен анализируемым, то систематич. погрешность может практически отсутствовать. В противном случае из-за указанныхвлияний на стадии испарения аэрозоля и атомизации возможны большие ошибкианализа. Существ. роль при этом играют и дисперсность аэрозоля и качествораспыляющего устройства.
с помощью образцов, содержащих точно известные кол-ва определяемого элемента. Если общий состав этих образцов идентичен анализируемым, то систематич. погрешность может практически отсутствовать. В противном случае из-за указанныхвлияний на стадии испарения аэрозоля и атомизации возможны большие ошибкианализа. Существ. роль при этом играют и дисперсность аэрозоля и качествораспыляющего устройства.ААА с пламенной атомизацией широко применяется в промышленности, медицине, экологии и др. наиб. успешно производится определение щелочных, щёлочноземельныхметаллов, серебра, меди, железа, марганца.
Существуют разл. методы с непламённой атомизацией (напр., с использованиемдугового, искрового, в т. ч. СВЧ-, разрядов). Однако наиб. распространениеполучил метод с электротермич. атомизацией проб (ЭТА). В этом методе атомизаторпредставляет собой трубчатую графитовую печь сопротивления, нагреваемуюв атмосфере аргона электрич. током. Раствор пробы вводится сквозь отверстиена внутр. стенку печи или графитовую пластинку внутри печи, где проба высушивается, проходит термообработку, и затем пары поступают в раскалённую полость печи. При такой обработке пробы атомизация происходит полностью.
Свет от ЛПК направляется вдоль оси графитовой трубки, проходит сквозьатомные пары и попадает на входную щель спектрофотометра. Интенсивности I Х и I0 регистрируются фотоэлектрич. приёмником. Благодарябыстрому разогреву печи на стадии атомизации, импульсному поступлению паровв зону поглощения света и малому объёму этой области мгновенная концентрацияатомов значительно выше, чем при пламенной атомизации. Если при этом используетсямалоинерционная регистрация поглощения, то пределы обнаружения элементоврезко (на 4-5 порядков) улучшаются. Поэтому метод ААА с электротермич. атомизацией особенно хорошо применять при определении микроколичеств. Так, напр., кадмий, цинк, медь, серебро с помощью этого метода регистрируютсяв кол-вах ~10-13-10-14 г; при массе пробы 0,001-0,005г это составляет 10-8-10-9%, что является рекорднымдля аналитич. методов. Кроме того, с помощью метода ААА возможен непосредственный(без растворения) анализ нек-рых веществ, однако при этом возникают трудностис градуировкой и несколько ухудшается воспроизводимость. Тем не менее методнашёл применение при определении примесей кремния, железа, кальция и т. п. в веществах высокой чистоты, что важно, напр., при контроле качестваматериалов для полупроводниковой техники, оптоэлектроники и др.
В ААА с электротермич. атомизацией кроме графитовых трубчатых печейиспользуют, напр., атомизаторы в виде вольфрамовой спирали. Они дают возможностьобнаружить мн. элементы, содержание к-рых в растворе 10-14-10-15 г. Совр. установки для ААА позволяют производить анализ (с погрешностьюне выше 0,05-0,1) в пробах, содержание определяемых элементов в к-рых ~10-5-10-7%.
Наиб. чувствительным С. а. является анализ с лазерным возбуждением спектра(для этого применяют перестраиваемые лазеры на красителях). Техника атомизациив этом случае мало отличается от используемой в ААА. Благодаря монохроматичностии высокой мощности излучения лазера возбуждается значительно большее числоатомов определяемого элемента, чем при термич. возбуждении. Чувствительностьобнаружения элементов при лазерном возбуждении чрезвычайно высока. Естьсведения, что удалось определять свинец в воде при содержаниях до
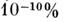 (1 пкг/мл).
(1 пкг/мл).Лит.: Зайдель А. Н., Основы спектрального анализа, М., 1965;Львов Б. В., Атомно-абсорбционный спектральный анализ, М., 1966; РусановА. К., Основы количественного спектрального анализа руд и минералов, 2изд., М., 1978; Спектральный анализ чистых веществ, Л., 1971; Лазернаяаналитическая спектроскопия, М., 1986. В. В. Недлер.
Молекулярный спектральный анализ
С помощью молекулярного С. а. (МСА) осуществляют качественное (идентификация)и количественное определения индивидуальных веществ или вещества в смесях. Это могут быть известное молекулярное вещество, новые стабильные и нестабильныемолекулы и частицы (ионы, радикалы и др.), разл. конформеры одних и техже молекул. Методом МСА исследуют вещества в любых агрегатных состояниях, растворах, плазме, адсорбц. слое и т. д. в широком диапазоне темп-р (отблизких к абс. нулю до сотен и тысяч градусов). Информативность методаопределяется строгой индивидуальностью спектров молекул, а сочетание методованализа по неск. видам спектров ещё более увеличивает надёжность определениясостава анализируемой пробы. Установлены общие закономерности, связывающиеспектры веществ с их строением.
Методы МСА основаны на сравнении измеренных молекулярных спектров исследуемогообразца со стандартными спектрами индивидуальных веществ (или расчётнымиспектрами, когда спектры индивидуальных соединений неизвестны). Используютвсе виды молекулярных спектров, характеризующих взаимодействие веществас эл.-магн. излучением (спектры поглощения, испускания, рассеяния, отражения, вращенияплоскости поляризации, фотоэлектронной эмиссии). Измерения могут производитьсяв широком диапазоне длин волн - от 10-12 м (
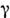 -излучение)до 103 м (радиоволны; диапазон частот 1018-106 Гц).
-излучение)до 103 м (радиоволны; диапазон частот 1018-106 Гц).Молекулярный спектр является однозначной характеристикой молекулы, определяетсяеё свойствами в целом, её структурой и свойствами входящих в неё атомов. В МСА используют электронные спектры (спектры поглощения в УФ- и видимойобластях, спектры люминесценции), колебат. спектры (ИК-спектры поглощенияи испускания, спектры комбинац. рассеяния), вращат. спектры (микроволновые),а также электронно-колебат. и колебательно-вращат. спектры и, кроме того, др.
виды спектров: рентгеновские (см. Рентгеноспектральный анализ),
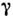 -спектры(см. Мёссбауэровская спектроскопия), фотоэлектронные спектры (см. Фотоэлектроннаяспектроскопия), спектры ядерного магнитного резонанса (ЯМР), электронногопарамагнитного резонанса (ЭПР), ядерного квадрупольного резонанса (ЯКР).
-спектры(см. Мёссбауэровская спектроскопия), фотоэлектронные спектры (см. Фотоэлектроннаяспектроскопия), спектры ядерного магнитного резонанса (ЯМР), электронногопарамагнитного резонанса (ЭПР), ядерного квадрупольного резонанса (ЯКР).Для целей МСА могут служить и др. методы исследований: для оптическиактивных молекул - дисперсия вращения плоскости поляризации, поляриметрия иэлектронный и колебательный круговой дихроизм (в УФ-, видимой иИК-областях, в спектрах КР). С появлением лазеров стали интенсивно развиватьсяметоды С. а., основанные на нелинейных эффектах, возникающих при взаимодействиивещества с лазерным излучением большой мощности; к ним относятся когерентноерассеяние света, вынужденное комбинац. рассеяние света (в т. ч. гиперкомбинац. рассеяние света, инверсное, усиленное поверхностью и др. виды комбинац. рассеяния света; см. также Нелинейная спектроскопия). ЧувствительностьМСА возросла как благодаря применению лазеров, так и за счёт использованияновых методов регистрации спектров (многоканальные методы, в первую очередьфурье-спектро-скопия, фотоакустич. спектроскопия) и применения низких температур(матричная изоляция, сверхзвуковые молекулярные пучки и др.). В нек-рыхслучаях МСА позволяет определять вещества в кол-вах до 10-12 г.
Качественный МСА позволяет по молекулярным спектрам идентифицироватьиндивидуальные вещества или устанавливать молекулярный состав исследуемогообразца. наиб. специфичны спектры веществ, содержащих в определ. интервалечастот исследуемого диапазона большое число спектрально разрешённых линийили полос (число полос во вращат. спектрах газообразных веществ в микроволновомдиапазоне достигает ~ 106).
Для повышения информативности МСА в нек-рых случаях измерение спектровкомбинируют с др. методами идентификации веществ, напр. сочетают ИК-спектрометри газовый хроматограф, что позволяет получать спектры индивидуальных компонентсложной смеси веществ. В связи с развитием фурье-спектроскопии, резко повысившейчувствительность ИК-спектрометров поглощения, стало возможным измерятьспектры отд. хроматографич. фракций при содержании исследуемого вещества~10-9 г. Сочетание ИК-спектрометров и спектрометров комбинац. рассеяния с микроскопом даёт возможность получать спектры микрообразцовразмером ~1 мкм и исследовать распределение веществ на поверхности гетерогенныхобразцов.
Разновидностью МСА является структурно-групповой анализ, позволяющийопределять в смеси не отдельные вещества, а классы веществ, имеющих общийспектральный признак, напр. органич. кислоты и кетоны. Метод основан наналичии в молекулярных спектрах т. н. характеристических частот. наиб. ярко это проявляется в колебат. спектрах. Напр., для всех нитрилов, содержащихгруппу
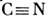 , в спектре появляется полоса в области 2200-2300см -1, для всехтиоспиртов с группой S - Н в спектре появляются полосы в области 2500-2600см -1, в спектрах всех органич. кислот имеются принадлежащиегруппе СООН полосы в области 1600-1750 см -1.
, в спектре появляется полоса в области 2200-2300см -1, для всехтиоспиртов с группой S - Н в спектре появляются полосы в области 2500-2600см -1, в спектрах всех органич. кислот имеются принадлежащиегруппе СООН полосы в области 1600-1750 см -1.Метод структурно-группового анализа позволяет определить класс, к к-ромупринадлежит вещество, и наличие тех или иных функциональных групп. Так, в промышленности применяется метод анализа нефтяных фракций на содержаниенепредельных углеводородов по спектрам комбинац. рассеяния света.
Качественный МСА производят путём сравнения получаемого спектра со стандартнымиспектрами. Созданы библиотеки, включающие десятки тысяч спектров. Анализсущественно ускоряется и упрощается при использовании ЭВМ, в память к-ройвводятся стандартные спектры. В ЭВМ сравнение может вестись как по всемуспектру, так и по отд. спектральным признакам, измеряемые спектры можновводить непосредственно в память ЭВМ. Если в библиотеке искомого спектранет, то спектр анализируемого вещества сопоставляют с теоретически рассчитанным. С помощью систем «искусств. интеллекта» рассчитывают колебат. спектры длянаиб. вероятных структур молекулы на основании заложенных в банк данныхсведений о эл.-оптич. и энергетич. параметрах молекул. Методами квантовойхимии рассчитывают электронные и колебат. спектры достаточно сложныхмолекул, к-рые также могут использоваться при идентификации веществ.
В науч. исследованиях часто проводят МСА неустойчивых и короткоживущихмолекул, а также анализ промежуточных продуктов хим. реакций и изучениеих кинетики. Для этой цели разработаны скоростные методы возбуждения ирегистрации спектров. Так, с помощью фурье-спектрометров получают ИК-спектрыза время до 10-3 с, при импульсном лазерном возбуждении - спектрыкомбинац. рассеяния за время ~10-9 с, спектры поглощения и флуоресценцииза время ~10-12 с и даже 10-15 с (см. Фемтосекунднаяспектроскопия).
При низких темп-pax время жизни неустойчивых молекул возрастает, чтопозволяет изучать их обычными спектральными методами. Одновременно за счётсужения линий, сопровождающегося ростом их пиковой интенсивности, а такжелучшего разрешения тонкой структуры существенно возрастают чувствительностьи информативность спектров. В т. н. методе матричной изоляции исследуютспектры разбавленных твёрдых растворов, когда исследуемое вещество заключенов твёрдой матрице инертного газа (Ne, Ar, Кг, Хе), азота и др. газов притемп-pax ок. 10 К; хорошо разрешённые узкие спектры вещества получают методоммолекулярных пучков, когда находящаяся под большим давлением смесь пароввещества и газа-носителя (обычно No, Аr) со сверхзвуковой скоростью вытекаетчерез узкое сопло, адиабатически охлаждается до темп-ры ниже 1 К и затемрегистрируются спектры. В этом случае могут быть спектроскопически идентифицированыдаже такие неустойчивые частицы, как ван-дер-ваальсовы молекулы.
Количественный МСА наиб. часто проводят по спектрам поглощения. В основеметода лежит Бугера - Ламберта- Вера закон:

где I0 и I - интенсивности падающего и прошедшего через образецизлучения, l - толщина слоя, с - концентрация вещества. Коэф. поглощения
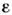 (молярный коэф. погашения) определяет поглощат. способность вещества начастоте излучения. Закон Бугера - Ламберта - Бера можно использовать вМСА только в отсутствие зависимости е от с, к-рая обычно связанас наличием в растворе межмолекулярных взаимодействий (напр., ассоциации).МСА по спектрам поглощения наиб. удобен для растворов и жидкостей; длятвёрдых веществ и газов такие измерения более сложны.
(молярный коэф. погашения) определяет поглощат. способность вещества начастоте излучения. Закон Бугера - Ламберта - Бера можно использовать вМСА только в отсутствие зависимости е от с, к-рая обычно связанас наличием в растворе межмолекулярных взаимодействий (напр., ассоциации).МСА по спектрам поглощения наиб. удобен для растворов и жидкостей; длятвёрдых веществ и газов такие измерения более сложны.На практике обычно измеряют оптическую плотность
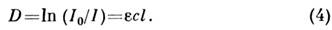
Если в смеси имеется п не реагирующих между собой веществ, тооптич. плотность на частоте v аддитивна:
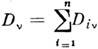
Это позволяет проводить полный или частичный анализ многокомпонентныхсмесей. При этом задача сводится к измерениям оптич. плотностей в . точкахспектра смеси и решению системы ур-ний:
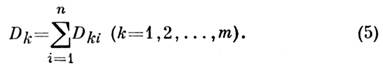
Необходимо знать величины коэф.
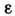 для каждой из компонент смеси при используемых значениях частот. Если соотношение(5) строго не выполняется, для проведения анализа смесей строят градуировочныекривые зависимости D от
для каждой из компонент смеси при используемых значениях частот. Если соотношение(5) строго не выполняется, для проведения анализа смесей строят градуировочныекривые зависимости D от 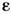 .
.Количественный МСА обычно производят с помощью спектрофотометров, измеряющихсоотношение
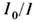 в широком диапазоне v. Если полоса поглощения исследуемого вещества изолированаи не перекрывается с др. полосами поглощения смеси, то анализ многокомпонентнойсмеси может осуществляться по этой полосе (как и для однокомпонентноговещества) по ур-нию (4). Полоса может быть выделена при получении спектрав спектрометре, однако проще и дешевле её выделять с помощью светофильтра. В промышленности используют специализиров. анализаторы, имеющие набор светофильтров.
в широком диапазоне v. Если полоса поглощения исследуемого вещества изолированаи не перекрывается с др. полосами поглощения смеси, то анализ многокомпонентнойсмеси может осуществляться по этой полосе (как и для однокомпонентноговещества) по ур-нию (4). Полоса может быть выделена при получении спектрав спектрометре, однако проще и дешевле её выделять с помощью светофильтра. В промышленности используют специализиров. анализаторы, имеющие набор светофильтров.Количественный МСА по спектрам испускания или комбинац. рассеяния светаосуществляют путём сравнения полученных спектров со спектрами эталонныхвеществ, записанными в тех же условиях. Интенсивность линии определяемоговещества сравнивают с интенсивностью нек-рой линии стандартного вещества(метод «внеш. стандарта») или с интенсивностью линии стандартного вещества, добавляемого к исследуемому в известном соотношении (метод «внутр. стандарта»).
Флуоресцентный МСА основан на сравнении спектров свечения раствора исследуемоговещества со свечением эталонных растворов близкой концентрации. Метод обладаетвысокой чувствительностью, но уступает методам поглощат. спектроскопиипо универсальности и избирательности. При использовании техники замороженныхрастворов (метод Шпольского; см. Шпольского эффект )информативностьспектров флуоресценции резко возрастает, т. к. в этих условиях спектрыобладают ярко выраженной индивидуальностью и резко различны даже для изомерови молекул близкого строения. Напр., метод Шпольского даёт возможность проведениякачеств. и количеств. анализа сложных смесей ароматич. углеводородов. Благодаряисключительно малой ширине спектральных линий в спектрах Шпольского удаётсядостигнуть пороговой чувствительности обнаружения нек-рых ароматич. веществ(~10-11 г/см 3).
Лит.: Беллами Л., Инфракрасные спектры сложных молекул, пер. с англ., 2 изд., М., 1963; Юденфренд С., Флуоресцентный анализ в биологиии медицине, пер. с англ., М., 1965; Сильверстейн Р., Басслер Г., МоррилТ., Спектрометрическая идентификация органических соединений, пер. с англ.,М., 1977; Э л я ш б е р г М. Е., Г р и б о в Л. А., Серов В. В., Молекулярныйспектральный анализ и ЭВМ, М., 1980; Смит А., Прикладная ИК-спектроскопия, пер. с англ., М., 1982; Вилков Л. В., П е н т и н Ю. А., Физические методыисследования в химии, т. 1-2, М., 1987-89. Б. В. Лакшин.
Физическая энциклопедия. В 5-ти томах. — М.: Советская энциклопедия. Главный редактор А. М. Прохоров. 1988.
.