INTERFACES
Une interface peut être définie, de la façon la plus générale, comme étant la zone qui sépare deux milieux A et B. Ces milieux doivent différer par au moins une des propriétés suivantes:
– composition chimique;
– nature des phases: solides, variétés cristallines, liquides, gaz;
– orientation cristalline (grains);
– ordonnancement (ordre-désordre);
– orientation du spin des électrons (domaines magnétiques).
En fait, l’usage est de parler de surface lorsqu’il s’agit de la limite entre un corps condensé (solide, liquide) et le vide ou éventuellement un gaz, et d’interface pour désigner la zone séparant deux milieux condensés.
À l’équilibre, le mouvement des atomes (ou leur changement d’état), de part et d’autre de l’interface n’est pas nul; mais il se compense statistiquement, de sorte que l’interface est immobile à l’échelle de l’observation. Dans un système hors d’équilibre, les échanges ne se compensent plus et l’interface se déplace. Si les cinétiques sont suffisamment lentes, l’interface peut sembler immobile: le système est métastable . C’est le cas pratiquement de toutes les interfaces limitant les solides, à la température ambiante.
L’épaisseur réelle de la zone de transition est extrêment faible: de un à quelques plans d’atomes, de sorte que l’interface peut être assimilée à une surface géométrique. Mais lorsqu’il y a variation de composition chimique, l’épaisseur de la zone perturbée, considérée comme interface, est généralement plus importante. L’interface continue cependant d’être assimilée à une surface tant que la méthode d’observation n’a pas une résolution latérale ou en profondeur supérieure à l’épaisseur réelle de l’interface. Examinée à une échelle plus fine, une interface peut soit disparaître, soit se résoudre en une succession d’interfaces élémentaires. Par exemple, la zone de diffusion entre deux métaux qui présentent une solution solide continue semble être une interface, quand l’échelle d’observation n’est pas suffisante. En revanche, si deux métaux forment un composé défini AB, le liseré du composé métallique AB apparaît comme une interface (macroscopique ou microscopique), alors qu’il est en réalité constitué de deux interfaces (microscopiques ou atomiques) A-AB et AB-B.
Une interface est conditionnée par les propriétés de chacun des deux milieux qu’elle sépare et elle est caractérisée par une énergie, dite interfaciale ou superficielle, qui dépend de la nature des liaisons atomiques au sein des deux milieux considérés. Les trois états de la matière (solide, liquide, gazeux) sont caractérisés par des types d’interaction entre atomes (ou molécules) très différents. À l’état solide, les atomes sont fortement liés entre eux, ce qui entraîne la rigidité du matériau et une forte énergie superficielle. Dans un liquide, la mobilité des atomes est plus grande, ce qui lui permet de conserver son volume mais non sa forme extérieure, et l’énergie superficielle est plus faible; pour un gaz, les interactions sont négligeables et cette énergie est nulle.
1. Énergies superficielles et interfaciales
Définition thermodynamique
La définition thermodynamique de l’interface a été souvent sujette à controverses. J. W. Gibbs l’a considérée comme une surface de division sans épaisseur, une surface mathématique, alors que J. D. Van der Walls et H. Bakker lui ont attribué une épaisseur faible mais finie. E. A. Guggenheim a introduit le concept de «phase superficielle», traitant l’interface comme une phase plane séparant deux phases homogènes. Considérons par exemple deux phases, un liquide et un gaz, et mettons-les en contact. On peut décrire le système comme étant la somme de trois régions: un liquide homogène, un gaz homogène, et une phase superficielle. Dans le concept de Guggenheim, on affecte aux phases superficielles les mêmes grandeurs extensives qu’aux phases volumiques mais avec un terme supplémentaire 塚d A, où 塚 est la tension superficielle de l’interface et A l’aire de la surface de contact des deux phases en présence. Pour augmenter l’aire de la surface de contact d’une quantité d A, il faut fournir un travail:
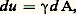
appelé énergie superficielle.
La tension superficielle 塚 correspond à une force qui s’exerce sur l’unité de longueur d’une ligne imaginaire découpant une surface, pour maintenir en contact les deux parties de la coupure. Cette force est tangente à la surface et perpendiculaire à la coupure. Elle s’exprime en newton par mètre alors que l’énergie superficielle s’exprime en joule par mètre carré. Tension et énergie superficielles ont donc les mêmes équations aux dimensions et sont souvent confondues. En fait, l’assimilation faite entre ces deux termes n’est pleinement justifiée que dans le cas des liquides en équilibre avec leur vapeur.
Physiquement, l’énergie superficielle a pour origine les forces exercées sur un atome par ses voisins. La résultante des forces d’attraction et de répulsion exercées entre deux atomes passe par un maximum pour une distance d entre ces atomes correspondant également au minimum d’énergie d’interaction et donc à l’équilibre. L’énergie qu’il faut fournir pour séparer les deux atomes est appelée énergie de liaison.
À l’intérieur d’un solide ou d’un liquide, l’ensemble des forces de liaison auxquelles est soumis un atome s’annulent alors qu’en surface il existe une résultante F dirigée vers l’intérieur (fig. 1). La surface se comporte donc comme si elle était soumise à une pression extérieure: c’est la pression superficielle . Cette image est assez parlante mais dangereuse, car elle ne traduit pas la réalité physique. L’énergie superficielle correspond aux énergies des liaisons qu’il a fallu rompre pour créer deux surfaces. Elle dépend du matériau (énergie de liaison) et elle est proportionnelle à l’aire de ces surfaces. Le principe de minimisation de l’énergie conduit donc à réduire cette aire.
D’une façon générale, en raison de la pesanteur, les liquides ont besoin d’être contenus dans un récipient solide. Il est alors nécessaire de considérer les forces d’interaction à l’interface solide-liquide. L’énergie superficielle 塚AB d’une interface A-B est la différence entre les énergies 塚A et 塚B qu’il a fallu fournir pour créer des surfaces libres dans A et dans B et l’énergie de liaison WAB récupérée lorsque les atomes de A et de B sont mis en contact. Elle est donnée par la relation de Dupré:

Lois générales
Loi de Laplace
Considérons dans un liquide une bulle de gaz de rayon r . Elle sera à l’équilibre lorsque la tendance à minimiser l’énergie de surface sera compensée par l’augmentation de pression à l’intérieur de la bulle. Le travail d W1 qui tend à contracter la bulle résulte de la pression externe (Pext) et de la tension superficielle 塚:
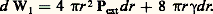
Le travail d W2 qui s’oppose à la contraction est dû à la pression interne (Pint):

Ces travaux s’annulent à l’équilibre. C’est l’équation de Laplace:


Loi de Jurin
Si l’on introduit un tube de verre propre et de faible rayon r dans de l’eau, le niveau de l’eau s’élève dans le tube et sa surface s’incurve pour former un ménisque (fig. 2 a). La surface du liquide est ainsi assimilée à la paroi d’une bulle d’air de rayon R et l’élévation du niveau du liquide d’une hauteur h compense la différence de pression de part et d’autre de cette paroi. La hauteur h est directement liée à la tension superficielle du liquide d’après la loi de Jurin:
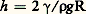
où 福 est la masse volumique du liquide et g l’accélération de la pesanteur. Si le mouillage (cf. infra ) n’est pas parfait ( 0), la loi de Jurin devient: h = 2 塚 cos / 福g R (fig. 2 a). S’il n’y a pas mouillage du tout, le niveau du liquide s’abaisse dans le tube (fig. 2 b).
Loi de Young
Si l’on considère trois liquides en équilibre au point triple, les angles de raccordement suivent la loi dite du triangle de Newmann. Si l’on ne considère alors que les seules forces de tension interfaciale, celles-ci s’annulent: 塚12 +塚23 +塚31 = 0. Dans le cas où une goutte de liquide L est déposée sur un solide S, il ne peut y avoir de distorsion de celui-ci et pour connaître l’angle de contact on applique généralement la formule de Young:

où 塚S est la tension superficielle du solide, 塚L celle du liquide et 塚SL la tension interfaciale entre le solide et le liquide (fig. 3). De façon plus générale, on fait intervenir la phase vapeur du liquide qui peut diminuer, par adsorption, l’énergie superficielle du solide. Celle-ci s’écrit alors 塚SV et la différence 塚SV 漣 塚S est appelée pression d’étalement 神e . Le coefficient d’étalement Se , qui traduit la mouillabilité, est la différence entre l’énergie d’adhésion (WSL) du liquide sur le solide et l’énergie de cohésion du liquide (WLL = 2 塚L). D’après les équations de Young et Dupré, le coefficient d’étalement s’écrit:
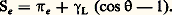
Valeurs des énergies superficielles et interfaciales
Les valeurs des énergies de surface sont délicates à déterminer en valeur absolue; la plupart des méthodes donnent l’énergie interfaciale entre un liquide et un solide, et ces valeurs sont très sensibles à de nombreux paramètres (cf. infra ). Dans la mesure du possible, il y a intérêt à se reporter aux tables de valeurs (cf. quelques exemples de valeurs dans le tableau), faute de quoi certaines méthodes permettent de les calculer ou du moins de les estimer.
Méthodes de calcul
La méthode la plus proche de la définition consiste à calculer le nombre de liaisons par unité de surface qu’il faut couper pour créer la surface libre, l’énergie de liaison d’un atome étant assimilée à la chaleur latente de vaporisation de celui-ci. Une autre méthode consiste à relier l’énergie de surface 塚 au travail de sortie des électrons (EW), par la relation:

où z est le nombre d’électrons libres de l’atome (valence), V le volume atomique et q un coefficient de structure qui tient compte de l’arrangement des atomes (q = 1, pour un métal liquide). Il existe d’autres formulations qui, bien que plus élaborées, ne conduisent pas à des valeurs plus satisfaisantes.
Pour les molécules complexes, on utilise une formule empirique qui est bien vérifiée expérimentalement, la formule du parachor :

où VL et VG sont les volumes molaires des phases liquide et gazeuse, et 神 une constante, appelée parachor, dont la particularité est d’avoir des propriétés additives. En effet, le parachor d’une molécule est la somme des parachors de chacun de ses éléments (atomes, cycles, doubles liaisons...).
Méthodes de mesure
Pour un solide, la mesure la plus directe de la tension superficielle est celle qui est obtenue par clivage . Encore faut-il que l’énergie ne serve qu’à rompre les liaisons du solide et qu’il n’y ait ni déformation ni échauffement. La méthode n’est donc valable que pour les matériaux fragiles. La méthode dite du fluage nul ne s’applique au contraire qu’à des températures élevées. La mesure du poids juste nécessaire pour compenser les forces de contraction dues à la tension superficielle et éviter toute déformation permet d’obtenir directement 塚. Cette méthode est particulièrement intéressante pour étudier l’influence de l’adsorption sur l’énergie superficielle des solides. Pour les liquides, on utilise généralement les phénomènes de capillarité ou d’équilibre de pression dans une bulle en appliquant les lois de Laplace ou de Jurin.
L’équation de Young, appliquée à l’équilibre d’une goutte de liquide sur un solide, conduit à l’énergie interfaciale liquide-solide par mesure de l’angle de contact, si l’on connaît les énergies superficielles de chaque phase (cf. supra , Loi de Young). D’autre part, lorsqu’un solide est chauffé à une température suffisamment élevée, un joint de grain arrivant en surface y creuse un sillon par diffusion superficielle ou par évaporation. Les mesures de profondeur et d’écartement de ce sillon permettent d’évaluer l’énergie interfaciale du joint.
Facteurs influençant l’énergie superficielle
La température
De façon générale, une élévation de température augmente l’agitation thermique et diminue l’énergie de liaison intermoléculaire, donc elle abaisse l’énergie superficielle. À la fusion, il y a une brusque diminution de cette énergie, puis on arrive à une température critique pour laquelle l’énergie superficielle s’annule: les forces de cohésion disparaissent et la substance ne peut exister qu’à l’état gazeux.
Rappelons qu’au cours du chauffage de substances, même considérées comme très pures, certaines impuretés ou constituants peuvent venir se concentrer en surface. C’est le phénomène de ségrégation qui se produit aussi bien aux surfaces qu’aux interfaces, dans les solides ou les liquides. La modification de composition chimique qui en résulte peut abaisser l’énergie superficielle de façon beaucoup plus importante que le simple effet de la température.
L’orientation cristalline
Nous avons vu que l’énergie superficielle dépendait du nombre de liaisons rompues autour d’un atome de surface. Ce nombre est évidemment fonction de l’arrangement cristallin. La variation de l’énergie superficielle en fonction de l’orientation cristalline est obtenue à partir du diagramme polaire de Wulff. L’anisotropie d’énergie superficielle est plus marquée pour les cristaux ioniques que pour les métaux et elle décroît par élévation de température.
Une conséquence de cette anisotropie d’énergie superficielle est le «facettage» de certaines surfaces métalliques. Une surface plane d’orientation quelconque peut se décomposer en une succession de plans d’indices plus simples et donc d’énergie superficielle plus faible (fig. 4).
La composition de surface
La thermodynamique montre qu’à l’équilibre l’adsorption d’atomes étrangers en surface entraîne toujours une diminution de l’énergie superficielle. Cette adsorption peut se produire par fixation de molécules provenant de l’atmosphère ou du milieu extérieur ou bien encore du cœur même du matériau; on dit dans ce dernier cas qu’il y a ségrégation. L’adsorption peut se limiter à une couche monomoléculaire, se poursuivre pour former un dépôt (métallisation d’une surface) ou un film mince de composé superficiel (oxydation des métaux).
Les substances qui ont la propriété d’abaisser l’énergie superficielle d’un substrat par adsorption sont appelées des agents tensio-actifs (cf. infra , Interface fluide-fluide ).
2. Les différents types d’interfaces
Rappelons qu’il n’y a pas d’interface lorsque les atomes ou les molécules ont tendance à se mélanger; c’est toujours le cas des gaz. Cependant, lorsque les gaz ont des densités très différentes, la décantation peut entraîner à forte échelle l’apparition d’une limite, mais il ne s’agit pas là d’une véritable interface puisque la composition varie de façon continue.
Interface corps condensé-gaz
Les échanges possibles entre un solide ou un liquide et un gaz sont la sublimation , l’évaporation et la condensation . La sublimation d’un solide ou l’évaporation d’un liquide donnent naissance à une phase gazeuse dont la pression à l’équilibre est appelée pression de vapeur saturante. Cet équilibre correspond, en fait, à un échange incessant de molécules entre chaque phase: il y a autant de molécules qui passent en phase gazeuse que de molécules qui se condensent. Leur nombre par unité de temps et de surface (n ) est égal au nombre de chocs de molécules gazeuses à l’interface; il se calcule par la théorie cinétique des gaz d’après la formule:

où Pv est la pression de vapeur en pascals, T la température en kelvins et M la masse molaire en kilogrammes. S’il n’y a pas condensation, cette formule permet de calculer la perte de molécules par évaporation. La pression de vapeur saturante croît avec la température. Sa loi de variation présente un changement de pente et non une discontinuité à la température de fusion.
Lorsque la phase gazeuse et la phase condensée sont de nature différente, il peut y avoir adsorption en surface, éventuellement suivie d’une dissolution en volume dont la cinétique est contrôlée par la vitesse de diffusion. À l’équilibre, la concentration du gaz en solution dans un liquide est, aux faibles teneurs, proportionnelle à la pression (loi de Henry). Dans le cas des solides, l’équilibre est régi par la dissociation des molécules en surface lors de l’adsorption: la solubilité d’un gaz diatomique est proportionnelle à la racine carrée de la pression (loi de Sievert).
Le principe de minimisation de l’énergie impose une surface lisse et de courbure régulière dès qu’il y a mobilité suffisante des atomes: c’est le cas des liquides. Cela ne se produit dans les solides que si la température est suffisamment élevée; le lissage de surface peut être obtenu par recuit à une température proche du point de fusion. Cependant, dans les solides cristallins, l’anisotropie d’énergie superficielle peut induire une rugosité d’équilibre due au développement de faces cristallines (striation, croissance des cristaux).
À l’échelle atomique, une surface quelconque se résout en faces simples avec formation de «marches» et de «crans». L’énergie superficielle n’est alors qu’une valeur moyenne, l’énergie propre de chaque atome croissant avec le nombre de liaisons rompues. Cela explique pourquoi l’interaction gaz-solide est beaucoup plus importante que l’interaction gaz-liquide, surtout lorsque le solide est rugueux et divisé (adsorption, catalyse). Deux autres modes de minimisation de l’énergie superficielle sont la relaxation et la reconstruction de surface. Dans la relaxation, seul le paramètre réticulaire perpendiculaire à l’interface est modifié.
Le phénomène de reconstruction correspond à un changement dans l’arrangement des atomes de surface. Il a lieu principalement dans les semi-conducteurs (Si), où l’existence de liaisons chimiques différentes ou insaturées peut créer des champs électriques susceptibles d’être ressentis jusqu’à 1 猪m de profondeur, alors que cette distance ne dépasse pas 1 nm dans les métaux.
Adsorption
Lorsqu’une molécule de gaz frappe une surface, elle peut s’y fixer car il n’y a pas saturation des atomes de surface. Selon la force de la liaison ainsi créée, on parlera d’adsorption physique ou d’adsorption chimique (fig. 5).
Typiquement, l’énergie de liaison est inférieure à 20 kJ/mole pour l’adsorption physique qui correspond à des forces de Van der Waals. Si la pression est suffisante, plusieurs couches monomoléculaires peuvent se physisorber les unes sur les autres. La physisorption est réversible: c’est la désorption qui est provoquée en abaissant la pression du gaz ou en élevant la température.
Lorsque les molécules ou les atomes adsorbés engagent de véritables liaisons chimiques avec le substrat, il y a modification de la distribution électronique de surface, on parle d’adsorption chimique. L’énergie d’interaction entre les molécules (Ec ) est alors beaucoup plus forte, elle peut atteindre 650 kJ/mole, et la distance interatomique est plus courte que précédemment. L’énergie mise en jeu fait intervenir une modification de la molécule gazeuse, par exemple sa dissociation (d’énergie Ed ). La chimisorption mettant en jeu des liaisons chimiques est spécifique du substrat et de l’adsorbat: elle conduit généralement à une couche monomoléculaire. La réaction peut se poursuivre soit par formation d’un composé tridimensionnel, soit par adsorption d’une nouvelle couche de gaz qui sera cette fois physisorbée.
Pour passer de la physisorption à la chimisorption, il faut généralement vaincre une énergie d’activation (Ea ) correspondant à la dissociation de la molécule à l’état adsorbé (fig. 5). Cette dissociation ne se produit spontanément que si la température du substrat est suffisante pour communiquer aux molécules cette énergie Ea . Ce même résultat peut être atteint en dissociant au préalable les molécules ou, mieux, en les «excitant» avant ou après physisorption: la chimisorption peut ainsi s’opérer pour des températures plus basses du substrat. L’excitation est obtenue par formation de plasmas, par bombardement électronique ou par interaction avec un rayonnement. Cette dernière méthode présente l’intérêt d’être sélective si l’on choisit la longueur d’onde en résonance avec la liaison à briser (bande d’absorption). La rugosité de surface facilite également l’adsorption des gaz. Outre l’augmentation globale de l’aire de l’adsorbat, elle fournit des sites privilégiés où les atomes de surface possèdent davantage de liaisons libres.
L’adsorption est exploitée comme méthode de pompage. Il est possible de fixer des quantités très importantes de gaz sur des zéolites (tamis moléculaires) refroidis à l’azote liquide. Le phénomène est réversible et les pompes sont régénérées par retour à la température ambiante. Les zéolites présentent une certaine sélectivité en fonction du diamètre des pores (masques à gaz, traitement des eaux de consommation...).
Désorption
C’est le phénomène inverse de l’adsorption: les atomes présents en surface, mais non constitutifs du substrat, passent dans la phase gazeuse. La désorption est d’autant plus facile que les «espèces superficielles» sont moins liées: ce sont les molécules physisorbées qui désorbent en premier. Pour déplacer l’équilibre, il est possible d’agir soit sur la pression, soit sur la température. La désorption peut également être provoquée, à des températures plus basses, par excitation au moyen de particules telles que des électrons ou des photons: désorption stimulée. On effectue ainsi l’élimination des polluants organiques superficiels à l’aide du rayonnement ultraviolet des lampes à vapeur de mercure.
La désorption sous vide s’appelle «dégazage»: les gaz quittant les parois sont pompés mais retardent l’obtention du vide limite. La désorption est accélérée par étuvage, mais une élévation de température permet également la diffusion vers la surface de l’hydrogène dissous dans l’acier inoxydable constitutif de l’enceinte. Les matériaux utilisés en ultravide nécessitent donc des traitements de surface très soignés en ce qui concerne la rugosité et la propreté chimique.
Catalyse de contact
Une application directe de l’adsorption-désorption est la catalyse. Le catalyseur est une substance qui permet de réaliser une réaction thermodynamiquement possible mais dont la cinétique est trop lente. Il y a tout d’abord adsorption des produits de la réaction, ce qui modifie leur état électronique (dissociation par exemple) et favorise leur rencontre et leur réaction en surface. Il y a ensuite désorption des produits formés, donc régénération de la surface du catalyseur. Les catalyseurs sont essentiellement les métaux de transition (Pb, Cu, Ni), dont les électrons d peuvent former des liaisons de covalence, ou des oxydes non stoéchiométriques (oxydes de fer). Afin d’augmenter l’aire de contact et d’accroître le nombre de sites «actifs» (défauts), les catalyseurs sont utilisés sous forme dispersée: poudre, petits agrégats.
Interface fluide-fluide
Les interfaces liquide-liquide sont lisses et de courbure régulière. La condition d’existence d’une interface liquide-liquide est que chacun des fluides soit saturé en l’autre fluide. En effet, si deux liquides se comportent de telle façon que des molécules de l’un puissent se substituer à des molécules de l’autre sans que les forces d’interaction soient modifiées, les deux liquides sont miscibles en toute proportion et il n’y a pas d’interface. Mais si, pour chacun, les forces d’interaction d’une molécule avec ses voisines sont très différentes, alors il y a démixtion , donc interface.
La miscibilité ou la démixtion sont donc fonction des forces d’interaction entre les molécules. C’est pourquoi, le plus souvent, en élevant la température, donc en augmentant l’agitation thermique et en diminuant les forces d’interaction de chaque phase, on favorise la miscibilité. On comprend également que l’eau soit un très bon solvant puisque sa molécule, polaire, a des affinités pour toutes les autres molécules polaires si des problèmes de taille ne viennent s’interposer.
Puisque c’est aussi l’énergie d’interaction ion-dipole qui stabilise les ions en solution, si le solvant a une constante diélectrique très faible, le sel sera peu soluble (interaction ion-solvant faible); c’est ce qui explique la faible solubilité du benzène dans le tétrachlorure de carbone.
Quand l’aire des interfaces fluide-fluide est très grande, l’ensemble des deux phases s’appelle colloïdes. Ce sont de toutes petites particules (environ 500 nm de diamètre) d’une phase dispersée dans un fluide et dont la principale caractéristique est d’avoir une grande surface. Ces colloïdes sont soit des sols , dispersions de solides dans les liquides (par exemple: les peintures; les boues), soit des aérosols , dispersions de solides ou de liquides dans des gaz (par exemple: le brouillard, les laques cosmétiques, les fumées), soit encore des émulsions qui sont des dispersions de liquides dans des liquides (par exemple: le lait, la mayonnaise). Du fait de leur grande surface, les colloïdes sont thermodynamiquement instables (lois de Gibbs), et ce principe de minimisation de l’énergie de surface devrait transformer ces colloïdes en deux phases non miscibles. Pour éviter cette coalescence, il faut donc diminuer l’énergie interfaciale par l’utilisation de produits appelés agents de mouillage . Ce sont les stabilisants, émulsionnants, tensioactifs, détergents ou surfactants. Ces émulsions sont très utilisées industriellement, en particulier dans l’agroalimentaire ou la peinture.
Effets des agents de mouillage
Ils sont en général formés d’une chaîne hydrocarbonée, la queue, et d’une tête polaire ou ionique (fig. 6 a). La queue n’a pas d’affinité pour les molécules d’eau de l’environnement, elle est hydrophobe. La tête, par contre, agit fortement avec de l’eau (interaction dipole-dipole ou ion-dipole) et elle est solvatée: elle est hydrophile. C’est cette différence entre la tête et la queue qui confère au surfactant la propriété de s’adsorber à une interface donnée pour diminuer la tension superficielle. Par exemple, le produit tensioactif, s’il est introduit dans un mélange eau-huile, va se placer de telle sorte que les queues hydrophobes de ses molécules seront repoussées par le liquide alors que les têtes hydrophiles seront dans l’eau. Au repos, les liquides sont séparés par densité, leur interface est plane, et le détergent se fixe sur cette interface, la tête vers l’eau (fig. 6 b). Si l’on agite l’ensemble et que la concentration en détergent est suffisante (concentration micellaire critique), il y a formation de gouttes entourées de surfactant appelées micelles .
De façon générale, l’agent tensioactif se fixe à l’interface du côté du liquide qui le mouille le mieux. Dans le cas des savons, où la tête est large, l’émulsion est constituée de gouttes d’huile dans l’eau (fig. 6 c). De plus, s’il s’agit d’un savon de soude, la tête est ionisable (l’ion alcalin passe en solution) et ainsi les gouttes chargées négativement se repoussent, ce qui évite toute coalescence. En modifiant la nature et la taille de la tête ou de la queue des molécules, il peut se produire une inversion de l’émulsion: par exemple, les forces agissant sur les queues peuvent tendre à déformer l’interface jusqu’à la faire «se retourner», on obtient alors des gouttes d’eau dans l’huile (fig. 6 d).
Un corps tensioactif peut également conférer à un liquide un pouvoir mouillant vis-à-vis du solide s’il peut s’y lier. C’est ainsi qu’un détergent, dont la queue hydrophobe peut adhérer au tissu, va également se fixer sur les particules de graisse déposées sur celui-ci. Les têtes hydrophiles tendent à se repousser et ces particules de graisse peuvent ainsi se détacher des tissus: c’est le principe des lessives.
Les procédés de séparation par membrane liquide sont une application directe de ce type d’interaction. La séparation des produits du mélange s’effectue par diffusion sélective à travers la membrane organique sous l’effet d’un gradient de concentration entre les deux interfaces de la membrane. C’est ainsi que l’on sépare certains hydrocarbures, que l’on peut dessaler l’eau de mer et effectuer de l’extraction liquide-liquide en hydrométallurgie (extraction de chrome dans les résidus industriels).
Si de l’air barbote dans de l’eau, les bulles formées au sein du liquide crèvent en arrivant à la surface; mais, si l’eau contient un agent tensioactif, les bulles sont stables et s’amassent pour former une mousse. Ces mousses sont particulièrement stables dans le cas des savons qui donnent des films suffisamment rigides et résistants.
Les émulsions sont très utilisées industriellement, mais si dans certains cas on cherche à les stabiliser, il arrive qu’on veuille les éliminer, pour traiter les eaux usées par exemple. On réalise alors la coalescence des micelles en neutralisant leurs charges négatives par adjonction de cations (sels d’aluminium ou de fer): c’est la floculation ou coagulation.
Structure
Dans le cas d’une interface entre deux fluides, on pourra parler de structure s’il existe une orientation privilégiée de certaines molécules d’un fluide par rapport à l’interface. À l’état liquide, il s’agit presque essentiellement d’orientation dans une direction perpendiculaire à l’interface mais sans ordre dans les deux autres directions. D’une façon générale, l’agitation thermique interdit toute «structure» dans un fluide sauf pour les états smectiques et nématiques des liquides, qui constituent ce qu’on appelle les cristaux liquides [cf. CRISTAUX LIQUIDES].
Interface solide-liquide
Solidification
Lorsqu’on refroidit un liquide, la mobilité des atomes diminue. Cela peut s’effectuer progressivement, le liquide devient alors pâteux (sa viscosité augmente), puis solide. Il n’y a pas de discontinuité dans la transformation, et les molécules conservent à l’état solide le désordre de l’état liquide: c’est l’état amorphe ou vitreux. Il existe peu de matériaux amorphes à l’état naturel (obsidienne des volcans) car ils ne sont généralement pas stables. Le verre est facile à obtenir mais l’amorphisation des métaux exige des trempes ultrarapides. De façon générale, l’arrangement des atomes dans les solides obéit à un ordre rigoureux: c’est l’état cristallisé. L’anisotropie de l’énergie superficielle des solides cristallisés entraîne l’apparition de faces cristallines privilégiées lors de leur croissance.
Équilibre solide-liquide
Deux possibilités se présentent: ou bien il y a attraction du type force de Van der Waals, ou bien il existe des forces dues à la présence de charges à la surface du solide (effets polaires, effets du type acide-base de Lewis). Ce type d’interaction qui existe entre les surfaces et certains polymères en solution dans les liquides entraîne, au voisinage d’une surface solide, des variations de concentrations positive ou négative. L’épaisseur concernée par ces variations est de l’ordre de grandeur des molécules de polymères.
Ces interactions liquide-solide sont d’une importance capitale en tribologie: le lubrifiant constitue un film liquide entre deux pièces solides qui doivent glisser l’une sur l’autre sans contact. Le film doit donc assurer un mouillage parfait et se reformer immédiatement s’il a été déchiré par des forces de cisaillement trop importantes.
Lorsqu’un solide est très divisé, les forces d’interactions solide-air peuvent devenir plus fortes que la résultante des forces correspondant à l’interaction solide-liquide et au poids du solide. Dans ce cas, les particules de solide remontent à la surface du liquide: c’est le principe de la flottation utilisée pour la séparation de certains solides. Ainsi, dans le traitement des minerais de cuivre (Cu2S, CuFeS2, FeS) les grains riches en silicate de la gangue argileuse tombent au fond de la cuve alors que les grains riches en sulfure forment une mousse qui flotte en surface et que l’on récupère facilement. Pour rendre la flottation plus sélective, on ajoute parfois un agent hydrophobe.
Lorsqu’un solide est plongé dans un liquide, les atomes ou molécules de la surface du solide ont la possibilité de se dissoudre dans le liquide. Le liquide peut, pour sa part, s’adsorber sur le solide, former un composé avec lui ou s’y dissoudre. Il existe aussi un phénomène mixte, réversible, c’est l’échange d’ions entre le liquide et le solide.
L’adsorption d’un liquide est analogue à celle d’un gaz (cf. supra , Adsorption ): on retrouve la particularité qu’ont les atomes de se fixer préférentiellement aux points d’émergence des défauts. Inversement, la dissolution du solide peut conduire à la formation de figures d’attaque localisées sur les défauts, ce qui permet de localiser les dislocations ou les joints de grains. Certains réactifs appropriés donnent sur le cuivre et l’aluminium des figures d’attaque géométriques dont les faces renseignent sur l’orientation cristalline de la surface. Dans un échantillon polyphasé, il y a souvent sélectivité du réactif d’attaque.
Lorsque la dissolution du solide n’est pas souhaitée, et qu’elle est suffisamment lente, on l’appelle corrosion. Si le produit de la réaction est adhérent et qu’il protège le matériau contre l’action ultérieure du liquide, son épaisseur peut être limitée à quelques couches atomiques: il y a passivation. C’est le cas des aciers inoxydables ou du titane.
L’échange d’ions est un phénomène naturel (solutions des sols et racines des plantes) que l’on utilise par exemple pour adoucir l’eau trop calcaire. Dans ce cas, on remplace les ions Ca2+ par des ions Na+ fournis par une résine dite échangeuse d’ions selon la réaction:

Cette réaction étant réversible, on peut «régénérer» la résine en la soumettant à un flux de solution très riche en ions sodium.
Les ions H+ des milieux acides peuvent s’échanger avec certains cations du verre, et cela est mis à profit, par exemple, pour la mesure du pH grâce aux électrodes de verre. De façon générale, si le liquide est conducteur (électrolyte), les échanges se font de telle sorte que le solide libère des cations: c’est le principe des piles électriques, dans lesquelles le métal le plus électropositif se dissout.
Inversement, l’électrolyse permet d’effectuer des dépôts métalliques: c’est la galvanoplastie très utilisée industriellement pour le nickelage, le chromage, l’argenture, la dorure...
L’oxydation anodique est une technique électrolytique qui permet de faire croître à la surface des métaux, en particulier de l’aluminium, une couche d’oxyde protectrice.
Particularité d’une interface solide-liquide en apesanteur
La construction des satellites et la vie dans l’espace a posé de nombreux problèmes pour le conditionnement des liquides puisque seules subsistent la tension superficielle et les forces interfaciales. La figure 7 représente la position d’un liquide dans un réservoir sphérique, selon qu’il mouille ou non la paroi, en présence ou en l’absence de pesanteur. Aussi, pour l’alimentation des moteurs de satellite, l’O.N.E.R.A. a mis au point des réservoirs sphériques munis de bafles tronconiques, mouillés par le propergol, et situés près des orifices de pompage.
Interface solide-solide
Les atomes des solides cristallisés présentent un arrangement périodique qui peut se poursuivre jusqu’aux limites du solide. C’est le cas d’un monocristal. Mais il y a le plus souvent juxtaposition de plusieurs cristaux d’orientation différente appelés grains. L’interface de raccordement est le joint de grains. Certains corps purs (fer, étain...) et certains composés (silice, zircone...) présentent plusieurs variétés cristallines stables, chacune dans des domaines de température différents. Le passage de l’une à l’autre, qui s’effectue à température constante et s’accompagne de la formation d’une interface, s’appelle transformation allotropique . Les diagrammes de phases ou diagrammes d’équilibre sont la représentation des limites de composition des différentes phases, en fonction de la température, dans les mélanges de deux ou plusieurs éléments. Pour certaines compositions, il n’existe pas de phase homogène, mais nécessairement un mélange de phases.
Dans les solides cristallisés coexistent donc à la fois des joints de grains séparant deux orientations différentes de la même phase et des interphases où il y a discontinuité de composition chimique. Certains corps, de composition chimique différente, peuvent avoir des paramètres cristallins si proches que le réseau se poursuit sans discontinuité entre les deux phases: on dit qu’il y a épitaxie . Ce phénomène est bien connu dans le cas des aluns; il est mis à profit dans la réalisation des composants électroniques à l’aide de l’épitaxie par jet moléculaire ou M.B.E. (Molecular Beam Epitaxie).
Dans les solides amorphes (verres ou plastiques), les atomes ou les molécules ne présentent aucun ordonnancement entre eux. La seule interface possible est une discontinuité de composition chimique.
Joints de grains
Dans la zone de raccordement entre deux grains, certains atomes sont situés dans des sites qui correspondent aux positions normales pour chacun des deux réseaux: ce sont les sites de coïncidence. Les autres atomes se placent dans une position intermédiaire, la plus apte à minimiser l’énergie. La cohérence du joint s’exprime par la proportion d’atomes en sites de coïncidence. L’énergie interfaciale du joint de grain croît lorsque sa cohérence diminue. La cohérence est parfaite dans les joints de macle: les deux réseaux sont symétriques par rapport au plan d’interface. La cohérence est bonne dans les sous-joints ou joints de faible désorientation: les deux réseaux se raccordant par une succession de dislocations. La cohérence est faible pour les joints de forte désorientation. Lors de la solidification d’un bain fondu, des cristaux d’orientation différente prennent naissance (germes) et croissent jusqu’à se rencontrer pour former les joints de grains. La morphologie des joints est alors conditionnée par le gradient de température et par les vitesses de croissance; on a formation de dendrites lorsque l’anisotropie de croissance est forte. Les joints de grains prenant naissance en dernier, ils présentent souvent un très fort enrichissement en certaines impuretés: tel est le cas du soufre dans les aciers. Un autre mode d’élaboration des matériaux est le frittage (compaction d’une poudre suivi d’un recuit). Les joints de grains coïncident alors généralement avec la surface initiale de la poudre.
Les joints de grains ne sont pas stables thermiquement. Le recuit à une température suffisante provoque une minimisation d’énergie par diminution de l’aire des joints, c’est-à-dire par croissance des cristaux. Les joints de grains prennent alors un profil d’équilibre analogue à celui qu’on rencontre entre phases liquides. Pour obtenir à nouveau des grains de petite taille, il est nécessaire de recourir à une déformation importante du matériau (écrouissage) suivie d’un recuit à une température telle que la germination soit abondante (recristallisation).
Les propriétés tant mécaniques que chimiques des matériaux dépendent très fortement de la présence de joints de grains. La déformation d’un métal se fait par glissement le long de plans cristallographiques avec mouvement des dislocations. La discontinuité cristallographique des joints de grains constitue une barrière au mouvement des dislocations et donc à la déformation. Elle sera d’autant plus efficace que les joints seront plus nombreux et de plus faible cohérence. L’accumulation des dislocations contribue elle-même au durcissement. Les utilisateurs recherchent le plus souvent des matériaux à grains fins pour leurs meilleures propriétés mécaniques. À haute température, il existe cependant un autre type de déformation: le fluage aux joints de grains. Les grains glissent les uns sur les autres comme si les joints servaient de lubrifiant. Pour éviter ce phénomène, certaines aubes de turbine utilisées par l’aéronautique à températures très élevées sont monocristallines. Dans un certain nombre de cas (molybdène, tungstène...), la cohésion des joints de grains peut n’être pas suffisante pour résister aux contraintes engendrées par la déformation mécanique: on assiste alors à une rupture intergranulaire. L’énergie interfaciale des joints les rend plus sensibles à l’attaque par certains réactifs que la surface des grains eux-mêmes. Ce phénomène de corrosion intergranulaire est particulièrement pernicieux comme toute corrosion localisée.
Le phénomène de ségrégation des impuretés se produit parfois aux joints de grains; en effet, les impuretés en solution solide dans le matériau peuvent trouver des sites privilégiés dans les joints et s’y concentrer. Le facteur d’enrichissement est d’autant plus important que l’impureté est moins soluble et que le joint a une plus faible cohérence. La ségrégation est à l’origine de nombreuses précipitations intergranulaires. La présence d’impuretés ségrégées modifie l’énergie interfaciale (comme c’est le cas de l’adsorption pour l’énergie superficielle) de sorte que la tenue mécanique et la résistance à la corrosion des joints s’en trouvent affectées. La vitesse de diffusion des impuretés dans les joints est généralement beaucoup plus rapide qu’au sein du matériau. Il suffit de déposer une petite quantité de gallium en surface de l’aluminium pour qu’il diffuse très rapidement, dès la température ambiante, dans tous les joints de grains et provoque une décohésion totale de ceux-ci.
Interphases
La solidification de mélanges liquides donne souvent naissance à la juxtaposition de plusieurs phases avec les interfaces correspondantes que l’on appelle aussi interphases. Il en est de même lors du refroidissement de solutions solides homogènes sursaturées qui conduit à la précipitation de nouvelles phases. Le rôle des interphases devient essentiel dans le cas des eutectiques (à partir du liquide) et des eutectoïdes (à partir du solide), où les nouvelles phases se forment à l’état de fines lamelles alternées. La perlite des aciers est ainsi composée de ferrite (Fe 見) et de cémentite (Fe3C). Par chauffage à 700 0C, il y a minimisation de l’énergie interfaciale par globulisation de la perlite.
Il y a également formation d’une interphase lors de réactions superficielles conduisant à l’édification d’une couche de composé. C’est le cas de tous les métaux qui s’oxydent à l’air, seuls l’or et le platine y échappent. La croissance de la couche d’oxyde dépend de la vitesse de diffusion de l’oxygène ou des ions métalliques à travers elle, et donc de la température. Certains métaux tels que l’aluminium et le titane sont parmi les plus avides d’oxygène et sont en même temps ceux qui s’autoprotègent le mieux par suite de l’«imperméabilité» d’une très fine pellicule d’oxyde (quelques nanomètres). L’oxyde de fer n’apporte au contraire aucune protection, et il faut avoir recours à un revêtement externe: chromage, peinture...
L’emploi d’un revêtement externe permet de changer les propriétés superficielles des matériaux (protection, frottement, usure...) sans en modifier le comportement massif. Nous avons vu certains modes d’obtention de revêtement à partir de la phase vapeur (méthodes physique ou chimique) ou de la phase liquide (électrolyse), mais il existe bien d’autres techniques: immersion, pulvérisation, étalement... Les revêtements peuvent être constitués de métaux, de céramiques ou de polymères organiques.
Un matériau constitué par plusieurs phases intimement mélangées possède des propriétés massiques très différentes de celles de chacun de ses constituants. De très nombreux exemples existent dans la nature et l’homme a utilisé cette potentialité depuis longtemps. Mais depuis peu de temps le principe en a été exploité systématiquement dans la synthèse de nouveaux matériaux: les matériaux composites . Ainsi, la structure feuilletée a permis d’effectuer de grands progrès dans la fabrication des skis, et la structure fibreuse, en particulier avec des fibres de carbone, a connu de très grands développements: pneus à carcasse radiale, pales d’hélicoptère, perches de saut, raquettes de tennis...
Le collage des matériaux consiste à créer des interfaces à très haute résistance mécanique avec les deux surfaces à assembler. Il connaît lui aussi une très forte expansion grâce à l’apparition de nouveaux polymères (araldite, cyanolite...) et à une meilleure connaissance des préparations des surfaces. Le collage tend à remplacer la soudure et la brasure dans certaines applications (construction automobile, aviation).
Toutes les interphases ainsi réalisées (revêtements, composites, collages) ont profondément changé la technologie. Le point crucial reste leur tenue mécanique après élaboration, et plus encore après vieillissement: c’est le problème de l’adhérence. Il est d’autant plus difficile à maîtriser que le nombre de paramètres à contrôler est considérable et qu’il n’existe pas de tests quantitatifs réellement représentatifs. Une autre difficulté demeure, la tenue en température: l’interphase initialement réalisée ne doit pas se modifier par réaction ou diffusion des deux matériaux. Cet écueil peut généralement être évité en interposant une barrière de diffusion choisie pour son imperméabilité aux constituants des matériaux, par exemple une couche de nickel.
La notion d’interface apparaît de la façon la plus évidente dans le cas de la séparation entre solide et liquide, mais c’est au sein de l’état solide qu’elle prend toute son importance. Il n’est pratiquement pas de matériau ou de produit manufacturé qui ne doive ses propriétés mécaniques physiques ou même chimiques à ses surfaces internes. Par ailleurs, ce sont les progrès réalisés dans la connaissance et la mise en œuvre des interfaces qui ont permis un développement rapide de la science des matériaux dans des domaines tels que les «multicouches» et les matériaux composites.
Encyclopédie Universelle. 2012.