CHLORE
Le chlore (symbole Cl, numéro atomique 17) est un élément chimique de la famille des halogènes. Sa molécule Cl2 (le dichlore) est un gaz verdâtre plus lourd que l’air. Irritant et suffocant, il fut employé comme gaz de combat pendant la Première Guerre mondiale. Trop réactif pour exister à l’état libre, le chlore est très répandu dans la nature, dans les eaux salées et les gisements de sel notamment, à l’état d’halogénures. Il est aussi un constituant important de la matière vivante: le sérum sanguin renferme environ 7 grammes de chlorure de sodium par litre.
La production mondiale de chlore a atteint près de trente-sept millions de tonnes en 1989. Sa progression fut très rapide entre 1965 et 1975 mais a ensuite ralenti. L’industrie chimique consomme environ 80 p. 100 du chlore pour la préparation de solvants, de plomb tétraéthyle, de plastiques, de caoutchoucs synthétiques, d’insecticides. L’industrie papetière est un autre utilisateur important. La fabrication du chlore fournit simultanément de la soude et de l’hydrogène: chlore et soude constituent donc des coproduits dont, normalement, les demandes s’équilibrent; cependant, il y a parfois déséquilibre; c’est ainsi que des besoins accrus en soude nécessitent, depuis le début des années quatre-vingt-dix, l’obtention d’une certaine quantité de celle-ci par caustification du carbonate de sodium.
1. Propriétés
Le chlore, dont le tableau 1 présente les principales propriétés physiques, se situe dans la septième colonne du tableau de Mendeleïev. Possédant sept électrons périphériques, il a tendance à en capter un huitième pour former l’anion chlorure Cl-, en passant au degré d’oxydation 漣 1; mais, avec ses trois doublets libres, il peut également former diverses liaisons atomiques dans lesquelles ce degré est compris entre + 1 et + 7.
Sa pression de vapeur à 20 0C étant de 6,5 bar (1 bar = 105Pa), le chlore est aisément liquéfiable, soit par refroidissement au-dessous de 漣 35 0C, soit par compression sous 7 à 10 bars, soit par refroidissement et compression.
Il est assez soluble dans l’eau (4,6 l/l à 0 0C; 2,3 l/l à 20 0C) et peut donner un hydrate (voisin de Cl2 . 6H2O) vers 0 0C; il est soluble dans le tétrachlorure de carbone CCl4. Élément très actif, il est, du fait de son caractère électronégatif, très avide d’hydrogène et se comporte souvent comme un oxydant. Il réagit avec la plupart des éléments non métalliques, sauf avec le carbone; parfaitement sec, il est sans action sur les métaux, mais il suffit d’une trace d’eau pour qu’il les attaque tous, y compris l’or et le platine. Il est donc très corrosif.
Il déplace le brome et l’iode de leurs composés; il en est de même de l’oxygène, mais les réactions sont alors généralement limitées.
Ainsi, la réaction de dismutation:

est fortement déplacée vers la gauche, surtout à l’abri de la lumière, de telle sorte que l’on peut parler de solubilité du chlore dans l’eau. Sous l’action de la lumière, l’acide hypochloreux est décomposé avec libération d’oxygène et l’équilibre est déplacé vers la droite:

Cette réaction avec l’eau liquide explique les propriétés oxydantes de l’eau de chlore.
Avec les hydroxydes, le chlore donne lieu à des réactions de dismutation conduisant aux hypochlorites:

qui se transforment en chlorates:
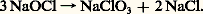
Le chlore donne lieu à des réactions d’addition avec certains anhydrides (obtention du chlorure de sulfuryle à partir de l’anhydride sulfureux: SO2 + Cl2S2Cl2) et avec les carbures éthyléniques, acétyléniques et benzéniques (préparation du dichloréthane, du tétrachloréthane et de l’hexachlorocyclohexane).
Il peut également fournir avec les composés organiques des réactions de déplacement ou de destruction totale et des réactions de substitution (chloroforme, tétrachlorure de carbone).
Le chlore peut être décelé par la coloration bleue qu’il communique à un papier imprégné d’iodiure de potassium amidonné, mais le fluor et le brome ont la même action. On le détecte mieux à l’aide d’un papier à l’orthotolidine (diméthyl-3,3 diamino-4,4 diphényle) qu’il fait virer du bleu au jaune.
2. État naturel et historique de la fabrication
Le chlore, sous forme de chlorures, est le plus abondant (0,2 p. 100 de l’écorce terrestre) des halogènes. L’évaporation de l’eau de mer fournit par litre 28,5 g de chlorure de sodium. 4,8 g de chlorure de magnésium et 0,8 g de chlorure de potassium. Les dépôts de sel gemme proviennent de l’évaporation d’anciennes mers; quand les espaces asséchés sont restés, durant l’évaporation, en relation avec le large, les sels les plus solubles ne se sont pas déposés et l’on trouve uniquement du chlorure de sodium avec du sulfate de calcium (gisement de Lorraine) ou éventuellement du chlorure de potassium (Alsace); lorsqu’il s’agit de l’évaporation d’une mer fermée à partir de laquelle tous les sels se sont déposés, les dépôts sont beaucoup plus complexes: on trouve alors des sels de potassium et de magnésium, comme à Stassfurt.
C’est le Suédois C. W. Scheele qui, dès 1774, découvrit le chlore par action de l’acide chlorhydrique sur le bioxyde de manganèse: on le considérait comme l’acide marin (HCl) déphlogistiqué, c’est-à-dire contenant de l’oxygène; ce n’est qu’après de longues discussions entre L. J. Gay-Lussac, L. J. Thenard et H. Davy qu’il fut défini comme un élément, auquel Davy donna le nom de chlore (1810), à cause de sa couleur verte (du grec諸福礼﨟, vert). L’histoire de la préparation industrielle, apparue au début du XIXe siècle, est marquée par quatre périodes:
1. Alors que le procédé Leblanc se développe, la découverte des chlorures décolorants par Berthollet conduit les soudières à intégrer à la fabrication du carbonate de sodium celle de ces nouveaux produits, afin d’éviter de répandre dans l’atmosphère des quantités de plus en plus grandes d’acide chlorhydrique résultant de la première phase du procédé Leblanc. Ce produit est transformé en chlore, d’abord par le procédé Scheele, amélioré par W. Weldon, puis, vers 1870, par le procédé Deacon utilisant la réaction équilibrée:
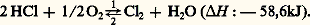
Cette réaction, exothermique dans le sens 1, exigeait d’opérer à la température minimale compatible avec une vitesse de réaction suffisante. À 450 0C, en présence de chlorure cuivrique comme catalyseur, on parvenait à transformer 47 p. 100 de l’acide chlorhydrique. La sévérité de la législation anglaise (Alkali Act de 1863) obligeant les soudières à ne plus déverser dans l’atmosphère l’acide chlorhydrique formé, donc à le transformer en chlore, explique l’importance prise en Grande-Bretagne par l’industrie des chlorures décolorants.
2. À partir du début du XXe siècle, l’électrolyse du chlorure de sodium en solution aqueuse, qui fournit du chlore à l’anode et de la soude et de l’hydrogène à la cathode, est venue concurrencer ce procédé et cela d’autant plus que, par suite du déclin du procédé Leblanc, on disposait de moins d’acide chlorhydrique résiduaire.
La mise au point industrielle de la liquéfaction du chlore en Allemagne avant la Première Guerre mondiale, en facilitant le transport du chlore, donc la fabrication des chlorures décolorants sur le lieu de leur utilisation, allait favoriser cette nouvelle technique.
En France, les chemins de fer s’étaient opposés au transport du chlore liquide. Ce n’est qu’en 1915, après que les Allemands eurent utilisé ce produit comme gaz de combat, qu’on développa l’électrolyse pour obtenir le chlore qu’exigeait la défense nationale.
3. Après 1918, la paix revenue, certains doutaient que les ateliers d’électrolyse ainsi créés puissent trouver la possibilité de placer leurs produits. Or, par suite du développement de la fabrication de la soie artificielle, les besoins en soude augmentèrent considérablement; simultanément, la production de chlore fut absorbée grâce aux demandes croissantes en solvants chlorés: tétrachlorure de carbone, trichloréthylène, perchloréthylène. Les craintes émises se révélèrent donc vaines, et cela d’autant plus que la découverte de plastiques chlorés tels que le chlorure de vinyle, puis le chlorure de vinylidène et le chloroprène, offrirent bientôt de nouveaux débouchés au chlore.
4. Les besoins en chlore devaient encore s’accroître avec l’utilisation de très nombreux autres produits chlorés: insecticides tels que l’hexachlorocyclohexane ou le D.D.T. (dichlorodiphényltrichloréthane), herbicides tels que l’acide 2,4-D (acide dichloro-2,4 phénoxyacétique), agents frigorigènes, tels que les carbures fluorochlorés, etc. Ainsi s’explique que, dans la plupart des pays industriels, la production du chlore se soit développée d’une façon étonnante, comme le montre la figure 1 en ce qui concerne la France, où elle a augmenté jusqu’en 1974; par la suite, elle a baissé puis remonté pour se stabiliser vers 1 300 000 tonnes par an. On tend en effet à substituer aux produits chlorofluorés des produits sans chlore, dits H.F.A.
3. Le problème de l’équilibre chlore-soude
Jusque vers 1950, les besoins en soude du marché étaient normalement couverts par la soude de caustification du carbonate de sodium et la soude électrolytique. La progression rapide de la production du chlore a alors fait craindre que l’on ne dispose de trop de soude, d’où la préoccupation de fabriquer du chlore sans soude. Ce «problème» a donné lieu à une très abondante littérature, à divers essais, et à quelques rares réalisations: deux usines aux États-Unis traitant, l’une du chlorure de sodium, l’autre du chlorure de potassium par l’acide nitrique afin d’obtenir du chlore et des nitrates, et deux usines électrolysant de l’acide chlorhydrique. Notons toutefois que la mise au point des procédés d’oxychloration à partir de l’acide chlorhydrique est sans doute une conséquence de cette préoccupation.
Indiquons aussi que, compte tenu de ce problème, on a fermé les ateliers de caustification, on a étendu les utilisations du carbonate de sodium en le substituant autant que faire se pouvait à la soude, et on a même préparé du carbonate de sodium par... carbonatation de la soude.
En 1966, la caustification était pratiquement arrêtée en France, et un équilibre entre chlore et soude s’est maintenu jusqu’en 1971, où est apparue une pénurie de soude.
On se trouve en fait devant deux coproduits dont les productions liées l’une à l’autre répondent sensiblement aux besoins du marché; des divergences peuvent toutefois apparaître et, s’il est aisé de répondre rapidement à une demande plus élevée en chlore, il n’est pas aussi facile de réaliser une surproduction de soude. Celle-ci constitue en fait le produit fatal car, si on peut la stocker, on ne peut pas stocker le chlore pour des raisons d’encombrement mais, surtout, de sécurité.
En France, 94,5 p. 100 du chlore produit proviennent de l’électrolyse de solutions aqueuses de NaCl, 3 p. 100 de solutions aqueuses de KCl et 2,5 p. 100 de l’électrolyse ignée de NaCl.
On doit également souligner que la fabrication de nombreux produits chlorés résulte très fréquemment de la chloration de composés carbonés, laquelle donne de l’acide chlorhydrique comme sous-produit. On est donc amené à développer de plus en plus l’obtention de chlore à partir de cet acide résiduaire, soit par électrolyse, soit de préférence par une adaptation originale de l’ancien procédé Deacon: l’oxychloration .
Les tentatives de modernisation de ce procédé avaient montré que, même en présence de catalyseurs actifs, il convenait de travailler à une température trop élevée pour que la réaction soit complète: d’où la nécessité de séparer le chlore de l’acide chlorhydrique et de recycler ce dernier.
En procédant à l’oxydation du chlorure d’hydrogène en présence d’éthylène qui fixe, sous forme de dichloréthane, le chlore formé extemporanément, on déplace complètement l’équilibre et la réaction devient complète; c’est le principe de l’oxychloration.
Appliquée initialement par F. Raschig pour préparer le chlorobenzène, produit intermédiaire de la fabrication du phénol, elle s’est développée de plus en plus pour obtenir le dichloroéthane à partir duquel on prépare le chlorure de vinyle par craquage:
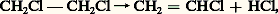
le chlorure d’hydrogène ainsi formé étant à nouveau traité par oxychloration.
Il convient de noter que le chlore produit dans les procédés d’oxychloration (dont le procédé «Chloé»), et qui est transformé in situ , sans être isolé, n’est pas inclus dans les statistiques de production de chlore.
4. Fabrication industrielle
On se limitera à indiquer le principe de sa préparation la plus importante: l’électrolyse de solutions aqueuses de chlorure de sodium, ou de potassium, et à décrire deux modes de réalisation.
Lors de l’électrolyse d’une solution de chlorure de sodium, tout se passe comme s’il se formait du chlore à l’anode et du sodium à la cathode, lequel en présence d’eau donne de l’hydrogène et de la soude. En l’absence de précautions spéciales, la soude et le chlore risquent de donner lieu, par suite de réactions secondaires, à la formation d’hypochlorite et de chlorate. Pour éviter ces réactions, on a recours à deux procédés: le procédé au diaphragme ou aux membranes et le procédé à cathode de mercure, dont les réactions peuvent être schématisées ainsi:
– procédé au diaphragme
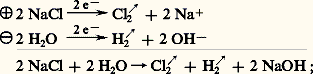
– procédé à cathode de mercure
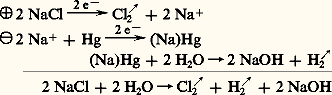
Procédé au diaphragme
Une couche d’amiante, parfois additionné d’une charge (sulfate de baryum, par exemple), montée sur une toile métallique ou une tôle perforée formant cathode, isole celle-ci de l’anode en s’opposant à la diffusion de la soude vers cette dernière: ce diaphragme, perméable aux ions, augmente cependant la résistance du bain et cela d’autant plus qu’il devient moins poreux par suite de la précipitation de chaux et de magnésie, consécutive à une purification incomplète des saumures.
Aux divers types de cellules classiques à diaphragme filtrant vertical, on a de plus en plus substitué les cellules Hooker, employées aux États-Unis depuis 1919 et introduites en France en 1945. Elles ne présentent pas l’inconvénient majeur inhérent à la plupart de ces appareils: un montage long et délicat.
La cellule Hooker de type S3 (fig. 2), qui a 1,5 m de largeur et 1,2 m de hauteur, comprend trois parties indépendantes: le fond portant les anodes, le porte-cathodes et le chapeau.
Le fond de la cuve est constitué par un bac en béton sur lequel est fixé par de l’asphalte le bloc anodique, qui comprend une barre d’arrivée de courant en cuivre et deux rangées de 15 anodes. Les anodes étaient initialement en graphite, encastrées dans un lit de plomb. Par suite de la formation de faibles quantités d’oxygène à l’anode, cette dernière était attaquée, avec formation d’oxyde de carbone souillant le chlore; les anodes s’usaient en se délitant et devaient être changées après une année de fonctionnement. On leur a substitué les anodes dites D.S.A. (dimensionnellement stables) en titane platiné (ou ruthénié), qui, théoriquement inusables, sont changées bien moins souvent (tous les dix ans et non plus tous les ans) que les anodes en graphite et ne donnent pas naissance à de l’oxyde de carbone.
Le porte-cathodes comporte un cadre s’adaptant sur le fond et deux peignes dont les dents, qui forment cathodes, s’intercalent entre les anodes. Ces dents comprennent une armature en tôle d’acier, dont la partie creuse constituera le compartiment cathodique d’où s’écoulera la soude et d’où se dégagera l’hydrogène, et sur laquelle est fixé le diaphragme. Pour fabriquer celui-ci, on plonge le bloc cathodique dans une suspension sodique d’amiante et on aspire sur le collecteur d’hydrogène, de façon à appliquer l’amiante sur la tôle d’acier. Le chapeau est mis en place et la cellule est prête.
La saumure à 315-320 grammes de NaCl par litre, dont on a éliminé la chaux et la magnésie par addition de soude, et éventuellement les sulfates par BaCl2, arrive en haut, par un diaphragme en tantale qui règle son débit et recouvre les électrodes portées à 80-85 0C par effet Joule.
Une telle cellule, qui peut fonctionner sous 30 000 ampères, travaille sous 3,45 V (tension théorique: 2,33 V) en donnant environ 900 kilogrammes de chlore par jour.
Un atelier comportant 4 boucles de 66 cellules chacune peut donc produire environ 250 tonnes de chlore par jour avec une consommation d’énergie de 2 720 kilowattheures par tonne, soit un rendement en courant (rapport entre la quantité de chlore préparé et la quantité théorique) de l’ordre de 95,5 p. 100 et un rendement en énergie (rapport de l’énergie utile pour l’électrolyse à l’énergie totale mise en jeu) d’environ 64 p. 100.
Le chlore, maintenu en très légère surpression dans la cellule, de façon à éviter un décollement du diaphragme, est aspiré par des pompes à anneau liquide; il contient environ 2 à 3 p. 100 d’impuretés (CO2: 1,6; CO: 0,1; H2: 0,22; 2 + 2: 0,2).
Les diaphragmes en amiante s’usent et doivent être changés tous les trois ou quatre mois. Pour cela, on ouvre la cellule, on extrait le porte-cathodes et on détruit le diaphragme par un arrosage à la lance, puis on en fabrique un nouveau.
Comme dans toutes les cuves, l’électrolyse n’est pas poursuivie jusqu’à ce que tout le chlorure de sodium soit décomposé car le rendement diminue à mesure que la concentration en sel baisse, tandis que la concentration en chlorate augmente. On extrait une solution contenant encore, par litre, de 160 à 165 grammes de sel, de 138 à 140 grammes de soude et 0,2 g de chlorate; cette solution est envoyée dans un atelier où on la concentre en continu dans des appareils à multiple effet: le chlorure de sodium précipite et on obtient de la soude à 50 p. 100 en poids renfermant moins de 1,5 g de chlorure par litre.
Certaines industries, comme celle de la rayonne, exigent des lessives exemptes de sel et il est nécessaire de faire subir à celles-ci un traitement spécial, soit en les mettant au contact d’ammoniaque, qui retient le chlorure de sodium (États-Unis), soit en faisant cristalliser la soude à l’état d’un hémiheptahydrate exempt de chlorure (Allemagne).
L’hydrogène, qui est maintenu en légère surpression pour éviter toute rentrée d’air, titre environ 99,8 p. 100.
Les cellules Diamond-Shamrock, également utilisées en France, fonctionnent comme les cellules Hooker.
Cellules à membrane
Depuis le début de la décennie 1970, on tente de remplacer les diaphragmes du type décrit plus haut par des membranes dites sélectives, ou même échangeuses d’ions. Comme les diaphragmes, ces membranes divisent la cellule en deux espaces, cathodique et anodique, et ne se laissent traverser que par les cations Na+; on peut ainsi obtenir une solution de soude, plus concentrée (de 30 à 35 p. 100) qu’avec les cellules à diaphragme, et ne contenant que de 0,02 à 0,2 p 100 de chlorure de sodium, ce qui évite l’opération d’élimination de ce sel; elles exigent une épuration poussée de la saumure qui doit contenir moins de 20 p.p.m. de Ca++, Mg++.
Ces membranes sont constituées par des hydrocarbures perfluorés tels que le tétrafluoroéthylène, mais comportant des groupes sulfoniques («Nafion», de Du Pont) ou carboxyliques (membranes de Asahi Glass Co.). Elles sont montées sur des cellules bi- ou monopolaires. Les cathodes sont encore en acier et les anodes en nickel.
Ne nécessitant que 2 600 kilowattheures par tonne de chlore, elles correspondent à une économie d’énergie par rapport aux cellules à diaphragme. Leur emploi est maintenant très répandu.
Cellules à cathode de mercure
On utilise une cathode circulante en mercure au contact de laquelle le sodium formé s’amalgame tandis que le chlore se dégage; le mercure contenant environ 0,2 p. 100 de sodium s’écoule de la cellule et passe dans un décomposeur dans lequel il fournit, en présence d’eau, de l’hydrogène et de la soude.
La cellule Solvay a été l’une des premières cellules utilisées; parmi les cellules récentes, nous citerons la cellule Pechiney, utilisée à Saint-Auban, les cellules de Nora et Mathieson, dont l’emploi s’est répandu aux États-Unis.
Les cellules Pechiney (fig. 3) sont constituées par un bac long de 12 mètres, de section rectangulaire (1,2 m 憐 0,30 m) en acier ébonité, légèrement incliné et sur le fond duquel s’écoule à une vitesse linéaire de 1 mètre par seconde, sur une épaisseur d’environ 0,2 mm, du mercure formant cathode. Une file d’anodes ayant la forme d’un T retourné sont fixées au couvercle de la cellule et peuvent être amenées par un dispositif adéquat à la distance voulue. Initialement en graphite, elles sont le plus fréquemment constituées par du titane recouvert d’oxyde de ruthénium.
La saumure, saturée en chlorure de sodium, doit être épurée du fait de l’instabilité des amalgames formés avec le magnésium et d’autres éléments. Elle s’écoule parallèlement au mercure et se trouve portée à environ 70 0C par effet Joule. Le sodium libéré par l’électrolyse du sel est entraîné à l’état d’amalgame et va au décomposeur.
Dans les anciennes cellules, le décomposeur était constitué par un bac disposé parallèlement au premier et incliné en sens inverse; les nouvelles cellules comportent un décomposeur vertical cylindrique rempli de morceaux de graphite, au sommet duquel une pompe envoie le mercure chargé d’amalgame. Au contact de l’eau et du graphite, l’amalgame se décompose avec formation de soude et d’hydrogène qui se dégage. La solution de soude, dont la concentration est réglée par un apport convenable d’eau, est pratiquement exempte de chlorure de sodium (0,1 g/l); on prépare ainsi des solutions à 50 et même à 70 p. 100 (avec un décomposeur à deux étages).
Le mercure retourne à l’électrolyseur, tandis que la saumure, dont la concentration a été amenée à 270 grammes par litre, est dirigée sur le saturateur en sel avant d’être recyclée.
Une telle cellule fonctionne sous 4,45 V et 90 000 ampères; elle fournit 3 tonnes de chlore par jour, avec une dépense de 3 300 kilowattheures par tonne. Les rendements en courant et en énergie sont respectivement de 96,5 et de 60 p. 100. On admet une consommation de 2,6 kg d’anode et de 300 grammes de mercure par tonne de chlore.
Pour une tonne de chlore on obtient 1,130 t de soude 100 p. 100 et 300 mètres cubes d’hydrogène. Le chlore obtenu est refroidi, afin de condenser l’eau entraînée, séché dans les laveurs à acide sulfurique, puis généralement liquéfié, ce qui élimine la majeure partie des impuretés qu’il contient.
Tandis que la lessive de soude, riche en sel, fournie par les cellules à diaphragme doit être concentrée, les décomposeurs des cellules à mercure fournissent directement une solution de soude de la concentration désirée.
L’hydrogène est séché par l’acide sulfurique puis généralement utilisé sur place dans les ateliers que comportent une usine de chlore pour les hydrogénations nécessaires aux fabrications annexes: cyclohexane, cyclohexanol, décaline, tétraline, etc. On prépare de moins en moins de chlorure d’hydrogène par synthèse, parce que de grandes quantités de ce produit résultent des chlorations.
De grandes usines de chlore ont employé cet hydrogène dans un atelier de synthèse d’ammoniac; cette utilisation est discutable car, étant donné les quantités d’hydrogène disponibles même dans une grosse unité de chlore comme Saint-Auban, l’atelier d’ammoniac qui produit de l’ordre de 40 tonnes par jour n’est pas à l’échelle des ateliers modernes d’ammoniac (1 000 t/j).
Comparaison des cellules à mercure et des cellules à diaphragme
La consommation d’énergie électrique par tonne de chlore produit est plus faible avec les cellules à diaphragme, mais un supplément d’énergie doit être fourni pour la concentration de la solution diluée de soude qu’elles fournissent, alors que les cellules à mercure produisent directement des solutions à la concentration voulue. Celles-ci ont également l’avantage de permettre l’obtention de soude pure, exempte de sel, telle qu’en exige la fabrication de la rayonne. Une légère perte en mercure résulte en revanche de l’entraînement par l’hydrogène et la soude. On leur reproche aussi leur encombrement et, surtout, l’immobilisation de mercure qu’elles nécessitent et, en conséquence, les investissements plus importants qu’elles exigent.
C’est pour pallier ce dernier inconvénient qu’on s’efforce depuis longtemps d’utiliser des cathodes tournantes à disques qui, en se recouvrant d’un mince film de mercure, permettent une économie importante de ce métal.
Les cellules à cathodes tournantes verticales, mises au point pendant la guerre, en Allemagne, ont pourtant été abandonnées parce qu’elles étaient peu pratiques; en revanche, les cellules de l’Asahi Glass Co. (Japon), utilisées au Japon et aux États-Unis, semblent donner satisfaction. Leur cathode est constituée par un disque horizontal en acier, tournant autour d’un axe vertical et qui, grâce à l’arrivée du mercure, se recouvre d’un mince film de métal (1 mm), lequel forme un amalgame, évacué ensuite. Une telle cellule fournit 2 tonnes de chlore par jour avec une dépense de 3 610 kilowattheures par tonne.
Un autre inconvénient des cellules à mercure est d’exiger de disposer de sel solide pour ressaturer la saumure venant du bac à électrolyse, alors que les cellules à diaphragme peuvent utiliser les saumures provenant des sondages. Or, dans les ateliers travaillant avec des cellules à diaphragme, la saumure provenant des cellules ne titre que 140 grammes de soude et de l’ordre de 160 grammes de sel par litre (cf. supra ); elle doit être concentrée dans des évaporateurs, où se dépose le sel. On voit donc l’intérêt du jumelage, soit dans une même usine (Lavéra), soit dans deux usines voisines (Pont-de-Claix et Jarrie) des deux modes de fabrication, l’atelier à diaphragme, qui utilise des saumures, ravitaillant en chlorure de sodium solide l’atelier au mercure. Ce couplage s’est considérablement développé.
En ce qui concerne la pollution, les cellules à diaphragme exigent quelques précautions quant à la manutention de l’amiante, du fait du risque de silicose. Les accidents de Minamata au Japon, résultant d’intoxication mercurielle, ont attiré l’attention sur le danger que pouvait présenter le rejet d’eaux résiduaires contenant du mercure et ont conduit à substituer peu à peu aux ateliers au mercure des ateliers à diaphragme. Ils ont également stoppé aux États-Unis la progression des cellules à mercure, qui tendaient à l’emporter sur les cellules à diaphragme. En revanche, en Allemagne, les cellules à mercure sont pratiquement les seules utilisées. En France, la production du chlore est assurée pour 54 p. 100 par des cellules à mercure, pour 44 p. 100 par des cellules à diaphragme et pour 2 p. 100 par électrolyse ignée du sel. La mise au point des cellules à membrane apportera certainement des changements importants dans cette répartition.
En France, le développement de l’industrie du chlore risquait d’être freiné par le coût de l’énergie, facteur important du prix de revient du chlore. Cela a conduit Progil à installer à Pont-de-Claix – avec l’accord d’É.D.F., qui détient le monopole de la distribution électrique – une centrale capable de fournir l’énergie nécessaire à cette usine, à un prix compétitif, grâce à la production combinée d’électricité et de vapeur. D’une puissance de 120 mégawatts, elle peut en effet produire annuellement de l’ordre de un milliard de kilowattheures et de 5 millions de tonnes de vapeur.
5. Utilisations
Le tableau 2 montre comment se répartissent en France les utilisations du chlore. Ces divers emplois suggèrent plusieurs remarques.
S’ils ont constitué le premier débouché du chlore, les chlorures décolorants – hypochlorites de sodium (eau de Javel) et de calcium (chlorure de chaux) –, obtenus par action du chlore sur les solutions de soude ou sur la chaux éteinte, sont depuis les années quarante en perte de vitesse. Au chlorure de chaux qui constituait éventuellement un vecteur du chlore on a substitué le chlore liquide. L’emploi des chlorazines, dérivés chlorés de l’acide cyanurique, se développe pour les laveries automatiques. Le chlorite de sodium est un agent de blanchiment important; mais on utilise surtout comme tel le chlore et le dioxyde de chlore.
Le chlore s’est également substitué aux chlorures décolorants pour le traitement des eaux d’alimentation ou des eaux résiduaires: le dioxyde de chlore, susceptible d’éliminer l’odeur et le goût désagréable dû aux composés phénoliques, devrait être de plus en plus employé.
Le rôle des dérivés chlorés comme anesthésiques (chloroforme, chlorure d’éthyle) est en régression mais le chloral, qui continue à être utilisé comme hypnotique, sert pour la fabrication du D.D.T.
De nombreux composés chlorés ont été employés pendant la Première Guerre mondiale comme gaz de combat; l’hexachloréthane, l’hexachlorobenzène et le chlorure de titane sont encore utilisés comme fumigènes.
Les dérivés chlorés jouent un grand rôle comme solvants: tétrachlorure de carbone, chloroforme, chlorure de méthylène, chlorure de méthyle et méthyl-chloroforme, mais surtout trichloréthylène et perchloréthylène, nécessaires le premier pour le dégraissage des métaux, le second pour le nettoyage à sec.
Des dérivés benzéniques et naphtaléniques sont également utilisés.
Les thioplastes préparés à partir du dichloréthane, puis d’éthers-oxydes dichlorés ont constitué les premiers caoutchoucs synthétiques; les Néoprènes résultant de la polymérisation du chloro-2 butadiène-1,3 (chloroprène) ont constitué des élastomères intéressants; la découverte des chlorures de polyvinyle puis celle du chlorure de vinylidène ont marqué l’histoire des plastiques.
L’oxychlorure de carbone (phosgène) intervient, comme produit intermédiaire, dans la fabrication des mousses d’uréthannes.
Les dérivés chlorés ont apporté une contribution importante dans la lutte contre les parasites: oxychlorure de cuivre, carbamates, obtenus par l’intermédiaire du phosgène, chloropicrine, puis D.D.T., hexachlorocyclohexane, dérivés de l’hexachlorocyclopentadiène (chlordane, heptachlore), et aussi comme herbicides sélectifs: acide 2,4 D (dichloro-2,4 phénoxyacétique), trichloroacétate de sodium, etc.
Le tétrachlorure de carbone et le chlorure de méthyle ont perdu leur rôle d’extincteur et d’agent frigorigène; ils sont remplacés par les dérivés fluorochlorocarbonés préparés à partir de dérivés chlorés et utilisés également comme propulseurs. Considérés comme susceptibles de polluer la haute atmosphère, ils font l’objet d’études pour être remplacés par des produits non chlorés, les carbures hydrofluorés dits H.F.A.
Les diphényles chlorés en mélange avec le diphényle constituent des lubrifiants et des diélectriques utilisés comme huiles de transformateurs.
Signalons le rôle essentiel du chlorure d’aluminium comme catalyseur et celui des dérivés chlorés comme intermédiaires pour la synthèse de produits très importants.
Enfin, à côté de l’emploi du chlore pour le désétamage du fer-blanc, insistons sur le rôle de cet élément en métallurgie: il fournit des chlorures métalliques volatils (on dit qu’il donne des ailes aux métaux): chlorures de titane, de zirconium, etc., qui servent de produits intermédiaires pour la préparation ou pour la purification de divers métaux.
chlore [ klɔr ] n. m.
• 1815; gr. khlôros « vert »
♦ Chim. Élément atomique (Cl; n° at. 17; m. at. 35,5), deuxième du groupe des halogènes, gaz jaune verdâtre, d'odeur suffocante. Le chlore est extrait du chlorure de sodium par électrolyse. Propriétés oxydantes, décolorantes, antiseptiques du chlore (cf. Eau de Javel). Le chlore est utilisé industriellement (fabrication du chlorure de chaux, des désinfectants, de l'acide chlorhydrique). — Composé hydrogéné du chlore : acide chlorhydrique (⇒aussi chlorure) . — Composés oxygénés du chlore : anhydride et acide hypochloreux (⇒aussi hypochlorite) , anhydride et acide chloreux (⇒aussi chlorite) , acide chlorique (⇒aussi chlorate) , acide perchlorique (⇒aussi perchlorate) .
⊗ HOM. Clore.
● chlore nom masculin (grec khlôros, jaune verdâtre) Corps simple, gaz de la famille des halogènes. (Élément chimique de symbole Cl.) Nombre atomique : 17 Masse atomique : 35,45 Densité par rapport à l'air : 2,5 Température d'ébullition : − 34,6 °C ● chlore (homonymes) nom masculin (grec khlôros, jaune verdâtre) clore verbe ● chlore nom féminin Herbe gentianacée des prairies, aux fleurs jaunes en cymes bipares, aux feuilles soudées par deux à la base.
chlore
n. m. CHIM élément non métallique, de la famille des halogènes (symbole Cl), de numéro atomique Z = 17.
— Gaz (Cl 2: dichlore) à l'odeur suffocante.
⇒CHLORE, subst. masc.
CHIM. Métalloïde gazeux, jaune verdâtre, d'odeur suffocante, de symbole Cl, appartenant à la famille des Halogènes. Eau de chlore. Solution aqueuse de chlore (cf. DUVAL 1959). Le linge blanchi au chlore (BLOY, Journal, 1899, p. 307). Les feuillages auxquels les lampadaires donnaient un vert de chlore (MONTHERLANT, Les Jeunes filles, 1936, p. 1065).
— P. méton. Composés du chlore, le plus souvent chlorure de chaux :
• 1. ... M. Bournisien aspergeait la chambre d'eau bénite et Homais jetait un peu de chlore par terre.
FLAUBERT, Madame Bovary, t. 2, 1857, p. 191.
• 2. ... les plus grands passaient leurs journées entières à polissonner au milieu des soldats, à dérober des poignées de riz ou de chlore aux sacs éventrés, ...
BARRÈS, La Colline inspirée, 1913, p. 299.
Prononc. et Orth. :[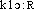 ]. Cf. lettre C, graph. ch. Ds Ac. 1835-1932. Étymol. et Hist. 1814 (NYSTEN). Empr. au gr.
]. Cf. lettre C, graph. ch. Ds Ac. 1835-1932. Étymol. et Hist. 1814 (NYSTEN). Empr. au gr.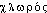 au sens de « vert, d'un vert jaunâtre ». Fréq. abs. littér. :48.
au sens de « vert, d'un vert jaunâtre ». Fréq. abs. littér. :48.
]. Cf. lettre C, graph. ch. Ds Ac. 1835-1932. Étymol. et Hist. 1814 (NYSTEN). Empr. au gr. au sens de « vert, d'un vert jaunâtre ». Fréq. abs. littér. :48.DÉR. 1. Chloré, ée, adj. a) Qui concerne le chlore. Odeur chlorée (H. PIÉRON, La Sensation, guide de vie, 1945, p. 193). b) Qui contient du chlore. Dérivés chlorés d'hydrocarbones (Hist. gén. des sc., t. 3, vol. 2, 1964, p. 442). — [ ]. — 1re attest. 1838 (Ac. Compl. 1842); de chlore, suff. -é. — Fréq. abs. littér. : 1. 2. Chloreux, euse, adj. Acide chloreux. Monoacide non isolé de formule HClO2 (cf. DUVAL 1959). — Dernière transcr. ds DG : klò-reú. — 1re attest. 1824 (NYSTEN); de chlore, suff. -eux. 3. Chloride, subst. masc., vieilli. Un des cinq métalloïdes ayant le chlore pour type. Synon. halogène (cf. DUVAL 1959). — Seules transcr. ds BESCH. 1845 et ds LITTRÉ : klo-ri-d'. — 1re attest. 1816 (L.-J. GAY-LUSSAC, Annales de chim. et de phys., t. 1, p. 171); de chlore, suff. -ide. 4. Chlorique, adj. a) Relatif au chlore. Acné chlorique (MACAIGNE, Précis d'hygiène, 1911, p. 307). b) Acide chlorique. Monoacide de formule HClO3 connu seulement en solution (cf. DUVAL 1959). — [
]. — 1re attest. 1838 (Ac. Compl. 1842); de chlore, suff. -é. — Fréq. abs. littér. : 1. 2. Chloreux, euse, adj. Acide chloreux. Monoacide non isolé de formule HClO2 (cf. DUVAL 1959). — Dernière transcr. ds DG : klò-reú. — 1re attest. 1824 (NYSTEN); de chlore, suff. -eux. 3. Chloride, subst. masc., vieilli. Un des cinq métalloïdes ayant le chlore pour type. Synon. halogène (cf. DUVAL 1959). — Seules transcr. ds BESCH. 1845 et ds LITTRÉ : klo-ri-d'. — 1re attest. 1816 (L.-J. GAY-LUSSAC, Annales de chim. et de phys., t. 1, p. 171); de chlore, suff. -ide. 4. Chlorique, adj. a) Relatif au chlore. Acné chlorique (MACAIGNE, Précis d'hygiène, 1911, p. 307). b) Acide chlorique. Monoacide de formule HClO3 connu seulement en solution (cf. DUVAL 1959). — [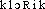 ]. — 1re attest. 1814 acide chlorique (NYSTEN, s.v. chlore); de chlore, suff. -ique. 5. Chlorite, subst. masc. Sel de l'acide chloreux (cf. supra dér. 2). Chlorite de sodium (cf. DUVAL 1959). — Dernière transcr. ds DG : klò-rit'. — 1re attest. 1831 chim. chlorite d'ammoniaque (E. Saubeiran ds Annales de chim. et de phys., t. 48, p. 141); de chlore, suff. -ite.
]. — 1re attest. 1814 acide chlorique (NYSTEN, s.v. chlore); de chlore, suff. -ique. 5. Chlorite, subst. masc. Sel de l'acide chloreux (cf. supra dér. 2). Chlorite de sodium (cf. DUVAL 1959). — Dernière transcr. ds DG : klò-rit'. — 1re attest. 1831 chim. chlorite d'ammoniaque (E. Saubeiran ds Annales de chim. et de phys., t. 48, p. 141); de chlore, suff. -ite.
]. — 1re attest. 1838 (Ac. Compl. 1842); de chlore, suff. -é. — Fréq. abs. littér. : 1. 2. Chloreux, euse, adj. Acide chloreux. Monoacide non isolé de formule HClO2 (cf. DUVAL 1959). — Dernière transcr. ds DG : klò-reú. — 1re attest. 1824 (NYSTEN); de chlore, suff. -eux. 3. Chloride, subst. masc., vieilli. Un des cinq métalloïdes ayant le chlore pour type. Synon. halogène (cf. DUVAL 1959). — Seules transcr. ds BESCH. 1845 et ds LITTRÉ : klo-ri-d'. — 1re attest. 1816 (L.-J. GAY-LUSSAC, Annales de chim. et de phys., t. 1, p. 171); de chlore, suff. -ide. 4. Chlorique, adj. a) Relatif au chlore. Acné chlorique (MACAIGNE, Précis d'hygiène, 1911, p. 307). b) Acide chlorique. Monoacide de formule HClO3 connu seulement en solution (cf. DUVAL 1959). — []. — 1re attest. 1814 acide chlorique (NYSTEN, s.v. chlore); de chlore, suff. -ique. 5. Chlorite, subst. masc. Sel de l'acide chloreux (cf. supra dér. 2). Chlorite de sodium (cf. DUVAL 1959). — Dernière transcr. ds DG : klò-rit'. — 1re attest. 1831 chim. chlorite d'ammoniaque (E. Saubeiran ds Annales de chim. et de phys., t. 48, p. 141); de chlore, suff. -ite.BBG. — PAMART (P.). De l'Alchim. à la chim. Vie Lang. 1969, p. 143.
chlore [klɔʀ] n. m.
ÉTYM. 1815; grec khlôros « vert, d'un jaune verdâtre ».
❖
♦ Chim. et cour. Corps simple, métalloïde (symb. : Cl; masse at. : 35,5; no at. 17), jaune verdâtre, d'odeur suffocante, dangereux à respirer, de densité 2,5, assez facile à liquéfier et assez soluble dans l'eau. || Le chlore ne se trouve pas dans la nature à l'état pur, on l'extrait en général du chlorure de sodium par électrolyse. — Propriétés oxydantes, décolorantes, antiseptiques du chlore. — Utilisation du chlore, des composés du chlore, comme narcotiques ou anesthésiants. ⇒ Anesthésie; chloral, chloroforme, éthyle (chlorure d'), méthyle (chlorure de); comme gaz asphyxiants (⇒ Chloropicrine, phosgène) et comme produits de blanchiment, de nettoyage, de désinfection. ⇒ Chlorure (chlorures décolorants), javel (eau de).
0 Il va assainir, purifier l'âme d'un mourant, quoi de plus simple ! C'est jeter du chlore dans une maison où vient de s'achever une maladie infectieuse.
M. Barrès, la Colline inspirée, XV, p. 254.
♦ Corps de la famille du chlore. ⇒ Halogène.
♦ Composé hydrogéné du chlore : acide chlorhydrique ou (vx) muriatique (ClH).
♦ Composés oxygénés du chlore : anhydride et acide hypochloreux (Cl2O et HClO; sels : hypochlorites), anhydride et acide chloreux (Cl2O3 et HClO2; sels : chlorites), peroxyde ou bioxyde de chlore, acide chlorique (HClO3; sels : chlorates), acide perchlorique (HClO4; sels : perchlorates).
❖
Encyclopédie Universelle. 2012.