HYPOPHYSE
L’hypophyse tire son nom de sa situation à la partie inférieure du cerveau. C’est une glande endocrine formée de deux parties distinctes et d’origine embryonnaire différente: le lobe postérieur et le lobe antérieur. Ce dernier est constitué de sept sortes de cellules que l’on peut caractériser par leurs réactions aux divers colorants et les techniques d’immuno-histochimie ultra-structurales. C’est donc un organe complexe par ses origines et sa structure; il l’est bien davantage par sa fonction physiologique et en particulier par ses relations avec les autres glandes, telles que la thyroïde, la corticosurrénale, les gonades et les glandes mammaires, traitées dans d’autres articles de cet ouvrage.
L’hypophyse joue un rôle dans le fonctionnement normal de l’organisme. En particulier, le lobe antérieur dirige la croissance de divers organes, et élabore des stimulines qui contrôlent la sécrétion des autres glandes endocrines. Aussi, conçoit-on que les lésions ou les troubles de son fonctionnement aient une importance capitale.
Ce contrôle qu’elle exerce sur de nombreuses glandes endocrines lui a valu le nom de « chef d’orchestre » endocrinien. En réalité, l’hypophyse n’est pas entièrement autonome; elle est dominée par une série d’actions nerveuses provenant de l’hypothalamus et du diencéphale. Celui-ci, relié par des connexions à tout l’ensemble du cerveau est une sorte de carrefour où viennent se rencontrer des influences organiques et toutes les réactions de la vie émotive qui, par son intermédiaire, retentissent sur l’hypophyse et ses sécrétions glandulaires. L’ensemble hypothalamus-hypophyse constitue un véritable système.
À la complexité anatomique de l’hypophyse, correspond une grande diversité de syndromes hypophysaires. Il peut arriver que le lobe postérieur ou le lobe antérieur, ou même, en ce qui concerne ce dernier, une seule catégorie de cellules sécrétrices, aient un fonctionnement anormal par suite de lésions diverses (tumeur, compression, traumatisme, infection), entraînant un hypo- ou un hyperfonctionnement de la glande. Parmi les syndromes d’hyperactivité les plus connus, on peut citer le gigantisme l’acromégalie et la maladie de Cushing. Les syndromes d’hypoactivité comprennent l’infantilo-nanisme, la maladie de Simmonds, le diabète insipide...
Au cours de cette étude, on se propose d’examiner la structure, l’origine et les fonctions de l’hypophyse, ainsi que les syndromes se rapportant au lobe antérieur et au lobe postérieur.
1. Structure, origine, fonctions
L’hypophyse, ou glande pituitaire, est un très petit organe, ayant chez l’homme à peine le volume d’une noisette, pesant environ 0,60 g. Reliée à la base du cerveau par la « tige pituitaire », elle est située à l’intérieur de la boîte crânienne, dans une dépression osseuse, la « selle turcique ». Bien qu’ayant l’apparence d’un organe unique, elle est formée, en réalité, de deux parties ou lobes, différant par leur structure, leur origine embryonnaire et leurs fonctions.
Le lobe antérieur
L’hypophyse se développe à partir de deux ébauches totalement différentes: une évagination de l’ectoblaste du stomodaeum située immédiatement en avant de la membrane pharyngienne et qui constitue la poche de Rathke , et un prolongement inférieur du plancher du diencéphale, l’infundibulum . Chez l’embryon de trois semaines, la poche de Rathke se dirige dorsalement à la rencontre de l’infundibulum. Vers la fin du deuxième mois, elle perd ses connexions avec la cavité buccale et se trouve en contact intime avec l’infundibulum. Au cours du développement ultérieur, les cellules de la paroi antérieure de la poche de Rathke prolifèrent activement pour former le lobe antérieur de l’hypophyse (lobe glandulaire ou antéhypophyse). Par la suite, une petite expansion de ce lobe, la pars tuberalis , se développe le long du pédicule infundibulaire pour venir finalement l’entourer complètement. La paroi postérieure de la poche donne la pars intermedia (fig. 1).
Le lobe antérieur a la structure caractéristique d’une glande à sécrétion interne, c’est-à-dire qu’il est dépourvu de canal excréteur et que ses produits de sécrétion se déversent directement dans le sang, au niveau du riche réseau de capillaires sinusoïdes disposé parmi ses éléments cellulaires. Ceux-ci étaient jadis classés grossièrement en cellules chromophobes, acidophiles (ou éosinophiles) et basophiles. Des méthodes fines de coloration et la microscopie électronique permettent aujourd’hui de distinguer sept types cellulaires, répondant aux sept hormones sécrétées par la glande [cf. HORMONES].
L’activité de ces cellules et la mise en circulation de leurs hormones sont sous la dépendance de centres nerveux situés à la base du cerveau, dans la région dite hypothalamus . Ces centres sont eux-mêmes réglés par les concentrations sanguines d’hormones, c’est-à-dire par les besoins de l’organisme (phénomène d’action en retour ou feed-back ). Ces relations se font par l’intermédiaire de substances chimiques, les releasing factors , sécrétées par des amas cellulaires spécialisés de l’hypothalamus, et qui gagnent l’antéhypophyse par le système porte hypophysaire (cf. HYPOTHALAMUS, fig. 3).
Des sept hormones sécrétées par l’antéhypophyse, quatre n’exercent leur action que par l’intermédiaire d’une autre glande endocrine, dont elles maintiennent la trophicité et l’activité sécrétoire. Telles sont la thyréostimuline (TSH ou thyréotrophine), la corticostimuline (ACTH ou corticotrophine), et les deux hormones gonadotropes (FSH et LH ou gonadotrophines) qui influencent respectivement la glande thyroïde, la glande corticosurrénale et les gonades (ovaires ou testicules). La prolactine (hormone lutéotrophique: LTH) tient sous sa dépendance la sécrétion lactée, tout en stimulant accessoirement l’ovaire.
Les deux dernières hormones hypophysaires agissent directement sur les tissus effecteurs; il s’agit de l’hormone de croissance ou hormone somatotrope (STH) et de l’intermédine ou hormone mélanotrope (MSH). La STH joue un rôle essentiel dans la croissance, non seulement du squelette, mais aussi de tous les tissus, et ceci grâce à de nombreuses modifications métaboliques: anabolisme des protéines, hyperglycémie, dégradation et utilisation des graisses. L’action physiologique de la MSH est beaucoup plus modeste car elle ne s’exerce que sur les cellules pigmentaires; elle se rattache au groupement ACTH [cf. HORMONES].
Le lobe postérieur
L’infundibulum donne naissance à la tige pituitaire et au lobe postérieur (ou lobe nerveux, posthypophyse, neurohypophyse ou pars nervosa ). Ce lobe est constitué par des fibres nerveuses dépourvues de myéline et par des cellules névrogliques. Les fibres émanent de neurones situés dans la région infundibulo-tubérienne ou hypothalamus et groupés en noyaux, dont les principaux sont les noyaux propres du tuber, les noyaux para-ventriculaires et supra-optiques (fig. 1 et 2).
Les hormones extraites de la posthypophyse (vasopressine et ocytocine) sont en réalité sécrétées par ces cellules nerveuses, et, cheminant le long de la tige pituitaire, gagnent le lobe postérieur, qui paraît constituer surtout un organe de stockage.
La vasopressine (ADH), hormone antidiurétique assure la réabsorption facultative de l’eau filtrée par les glomérules du rein, présidant ainsi à la régulation hydrique de l’organisme. Sa libération ou sa rétention sont commandées par les variations de la concentration saline du plasma et par celles du volume sanguin (cf. métabolisme HYDROMINÉRAL).
L’ocytocine , de structure voisine mais d’importance beaucoup moindre, intervient au niveau des éléments contractiles des muscles lisses de l’utérus, des vaisseaux sanguins et de la glande mammaire, participant ainsi aux mécanismes de la parturition et de l’éjection du lait.
La nature et les propriétés des différentes hormones hypophysaires ont été indiquées dans un autre article [cf. HORMONES]. On n’envisagera ici que les états pathologiques réalisés, chez l’homme, par leur défaut ou par leur excès, et qui sont, en somme, des aspects caricaturaux de leurs fonctions.
2. Syndromes antéhypophysaires
Ces syndromes sont complexes et variés, en raison de la multiplicité des fonctions de l’hypophyse et du retentissement des perturbations hypophysaires sur les glandes soumises à l’action de ses stimulines. Par ailleurs, le siège intracrânien de la glande, au contact du chiasma optique et au voisinage de la base du cerveau, explique l’intrication fréquente des troubles endocriniens avec des syndromes neurologiques primitifs ou secondaires.
Acromégalie et gigantisme
L’acromégalie et le gigantisme sont tous deux l’expression d’un hyperéosinophilisme hypophysaire , c’est-à-dire d’une sécrétion excessive d’hormone de croissance par les cellules éosinophiles classiques de l’antéhypophyse. La différence entre ces deux syndromes tient à l’âge auquel commence à se manifester l’hyperactivité hormonale: avant ou après la puberté. Ils peuvent d’ailleurs se combiner.
L’acromégalie
L’acromégalie, décrite en 1886 par Pierre Marie, est pratiquement toujours liée à une tumeur, le plus souvent bénigne (adénome), exceptionnellement maligne (carcinome), développée aux dépens des cellules éosinophiles. Aux signes proprement endocriniens, se joignent tôt ou tard des signes neurologiques dus à la compression des organes nerveux voisins. La maladie frappe des sujets jeunes, de vingt à quarante ans, de façon insidieuse et très lentement progressive.
Les signes endocriniens sont surtout d’ordre morphologique. C’est à eux que la maladie doit son nom (akros : saillie, extrémité; mégas : grand). L’excès d’hormone somatotrope entraîne une reprise de la croissance de tous les tissus, et principalement du squelette. Mais, la soudure des cartilages de conjugaison étant achevée, le sujet ne peut plus grandir. Il s’accroît en épaisseur et se déforme, l’hypertrophie étant surtout apparente aux extrémités.
Les mains épaisses et larges, prennent un aspect « en bêche », « en battoir ». Les paumes sont comme « capitonnées ». Le malade doit augmenter la pointure de ses gants, de ses souliers, de son chapeau. Le faciès subit des modifications caractéristiques: allongement antéro-postérieur du crâne; développement excessif des arcades sourcilières; épaississement du nez, des pommettes; allongement du menton avec projection en avant de la mâchoire inférieure. L’épaississement des narines, des lèvres, des oreilles et surtout de la langue (macroglossie) est manifeste. Tout le squelette participe au processus: les clavicules et les côtes sont épaissies et proéminentes; la saillie antérieure du sternum et le dos arrondi évoquent l’aspect du polichinelle. L’hypertrophie du larynx modifie le timbre de la voix. Ces déformations se font généralement sans douleurs; il existe pourtant des formes « rhumatoïdes ». Tous les viscères sont hypertrophiés (splanchnomégalie): le foie, les reins, le cœur. Les radiographies du squelette montrent l’épaississement des os, et souvent, au voisinage des articulations, des productions bourgeonnantes « ostéophytiques ».
À ces symptômes morphologiques, se joignent des troubles métaboliques résultant directement ou indirectement de l’excès d’hormone somatotrope: élévation du phosphore minéral du sang; diminution de l’excrétion urinaire des phosphates; augmentation du sucre sanguin, allant dans presque la moitié des cas non traités jusqu’à un véritable diabète; élévation du métabolisme basal par augmentation directe des oxydations cellulaires. La suppression des règles, fréquente chez la femme, est accompagnée d’un certain degré de virilisation. À une phase avancée, une impuissance sexuelle apparaît chez l’homme. Dans les deux sexes, on peut noter un écoulement anormal de lait.
Le tableau clinique est assez caractéristique pour permettre de reconnaître l’acromégalie et de la rattacher à sa cause (tumeur hypophysaire dérivée des cellules éosinophiles). Mais il n’est pas sans intérêt de mettre en évidence directement l’excès d’hormone par son dosage dans le sang, au moyen d’une méthode radio-immunologique. Normalement, la sécrétion de l’hormone est freinée par l’administration de glucose. En cas de tumeur, le freinage ne s’exerce plus. Des dosages répétés permettront ensuite de contrôler les effets des traitements mis en œuvre.
Les signes de tumeur hypophysaire sont habituellement plus tardifs. Annoncés par des céphalées tenaces, ils se caractérisent objectivement par un affaiblissement ou une perte de la vue dans la moitié externe du champ visuel de chaque œil (hémianopsie bitemporale, due à la compression du chiasma optique et pouvant aller jusqu’à l’atrophie optique avec cecité). Les radiographies du crâne montrent une selle turcique, très agrandie dans toutes les dimensions « ballonnisée », avec usure de ses parois, associée à une hypertrophie des apophyses clinoïdes antérieures.
La maladie évolue avec une extrême lenteur, ne représentant d’abord qu’une disgrâce, mais devenant à la longue une infirmité grave. Non traitée, elle conduit à la mort, à un âge plus ou moins avancé, soit par une complication du diabète, soit par suite de la compression cérébrale exercée par la tumeur, soit, plus souvent, à l’occasion d’accidents vasculaires cérébraux ou coronariens dus à une artériosclérose précoce avec hypertension artérielle.
Dans les formes peu avancées, sans troubles oculaires importants ni diabète, on recourt à des irradiations transcrâniennes de la tumeur, de préférence au moyen de la télécobaltothérapie « de mouvement ». En cas d’échec, on peut pratiquer une implantation de corps radio-actifs, tels que l’yttrium 90, à l’intérieur de la selle turcique. Mais, quand il existe des troubles oculaires menaçants, il est préférable d’enlever chirurgicalement la tumeur ou de la détruire par électrocoagulation ou cryothérapie, en l’abordant soit par voie transcrânienne sous-frontale, soit par voie nasale transsphénoïdale, beaucoup moins traumatisante. Le choix entre les deux méthodes dépend du mode d’extension de la tumeur, que précisent des radiographies après injection sous-arachnoïdienne d’air (pneumo-encéphalographie).
Le gigantisme
Le gigantisme par excès d’hormone de croissance hypophysaire est beaucoup plus rare; il survient chez des enfants ou des adolescents. Un sujet peut avoir une très grande taille (supérieure même à deux mètres), mais sans dysharmonie morphologique et sans troubles métaboliques. Le gigantisme pathologique, dont il est ici question, comporte un déséquilibre entre l’excès de développement statural et l’ensemble des fonctions organiques. On en distingue deux formes principales.
Dans le gigantisme avec infantilisme , les sujets ne grandissent pas seulement trop et trop vite, mais ils continuent encore à grandir bien au-delà de l’âge normal, tandis que leurs organes génitaux restent infantiles et que leurs caractères sexuels secondaires ne se développent pas. Ils présentent des membres démesurément longs, un buste étroit. D’intelligence fruste, ils sont impuissants et stériles, et succombent généralement avant l’âge de trente ans. Il existe, dans certains cas, des signes de tumeur hypophysaire; mais le fait est loin d’être constant. Le dosage d’hormone somatotrope dans le sang, avec épreuve de freinage par hyperglycémie provoquée, est alors indispensable pour affirmer que le gigantisme est bien lié à un excès de sécrétion de cette hormone.
Le gigantisme acromégalique se caractérise par le fait que la puberté finit par apparaître, amenant la soudure des cartilages de conjugaison. Le sujet cesse alors de grandir, mais on voit survenir des déformations de type acromégalique. Dans ces cas, l’existence d’une tumeur hypophysaire est plus fréquente et l’apparition d’un diabète est possible.
Le traitement est comparable à celui de l’acromégalie. En l’absence de signes de tumeur hypophysaire, on se contente d’irradiations de la glande. Mais, dès l’âge de douze à treize ans, il faut s’efforcer de déclencher la puberté par des séries d’injection d’hormones gonadotropes, ou même, si celles-ci sont insuffisamment efficaces, recourir à un traitement substitutif par les hormones sexuelles: testostérone chez les garçons, œstrogènes et progestatifs chez les filles.
Maladie de Cushing
Le syndrome de Cushing est une affection qui traduit une sécrétion excessive d’hormones corticosurrénales. Il possède un aspect clinique bien caractéristique et une physiopathologie définie, mais il peut résulter, en réalité, de causes différentes. Dans certains cas, qui ne sont pas les plus fréquents, il a pour origine une tumeur corticosurrénale bénigne (adénome) ou maligne (carcinome); et il ne s’agit donc pas d’une maladie d’origine hypophysaire. L’excès d’hormones corticosurrénales produit par la tumeur a même pour effet d’inhiber, par un phénomène de feed-back négatif, la sécrétion d’hormone corticotrope par l’hypophyse; il provoque aussi des altérations des cellules basophiles de l’hypophyse, mais qui sont secondaires et non causales.
Plus souvent, le même tableau clinique, traduisant également un excès d’hormones corticosurrénales, résulte d’une sécrétion excessive d’hormone corticotrope antéhypophysaire (ACTH). La maladie est bien alors d’origine hypophysaire, l’hyperactivité surrénalienne, liée à une hyperplasie bilatérale des capsules surrénales, n’étant qu’un phénomène secondaire. C’est à ces cas que l’on réserve le nom de maladie de Cushing.
La nature du dérèglement hypophysaire est elle-même variable. Il peut s’agir d’une tumeur de l’hypophyse, développée aux dépens des cellules « corticotropes » de la glande (J. Decourt et M. Herlant; J. F. Foncin et J. Le Beau, 1963). Cette éventualité n’est pas la plus fréquente. L’hyperactivité corticotrope de l’hypophyse reste le plus souvent de nature indéterminée, probablement d’origine supra-hypophysaire, par dérèglement hypothalamique et surproduction continue de CRF (cortico-releasing factor ).
La maladie s’observe surtout chez les sujets jeunes, de dix-huit à quarante ans, plus souvent chez la femme que chez l’homme. Elle n’est pas exceptionnelle chez l’enfant. Elle réalise un tableau complexe, formé d’une série de symptômes, dont aucun n’est à lui seul caractéristique, mais qui valent par leur groupement. Ce sont, par ordre de fréquence: un engraissement, portant surtout sur la face, le cou, le tronc, la racine des membres, épargnant les extrémités; une rougeur violacée des téguments, surtout du visage; des vergetures pourpres, rappelant celles de la grossesse; un certain degré d’hirsutisme chez la femme, associé à de la séborrhée, à de l’acné; des troubles génitaux tels que l’aménorrhée chez la femme (absence de règles, en dehors de l’état de grossesse) et l’impuissance sexuelle chez l’homme; une atrophie musculaire, plus ou moins masquée par l’adipose; une décalcification du squelette, surtout appréciable au niveau des vertèbres; une hypertension artérielle; des troubles de la glycorégulation pouvant aller jusqu’à un véritable diabète; souvent, enfin, des troubles psychiques.
Ces symptômes ne sont pas toujours au complet. L’association de quelques-uns d’entre eux doit faire suspecter l’hypercorticisme surrénalien et rechercher les signes biologiques. Ce sont essentiellement: l’excès du cortisol dans le sang, et la présence dans les urines de quantités élevées des métabolites dérivés des hormones corticosurrénales (17-cétostéroïdes et 17-hydroxycorticostéroïdes). En outre, et c’est le fait le plus caractéristique, l’administration de dérivés du cortisol, tels que la dexaméthasone, se montre incapable de réduire notablement ces métabolites en excès, c’est-à-dire de freiner la sécrétion hypophysaire d’hormone corticotrope.
La maladie étant identifiée, il convient encore de s’assurer qu’il ne s’agit pas d’une tumeur surrénalienne car, dans ce cas, le traitement comporterait l’ablation chirurgicale de cette tumeur, pratiquement toujours unilatérale. On recourt, pour établir ce diagnostic, à la radiographie des surrénales en s’aidant d’une injection d’air dans le péritoine, ou, mieux, en faisant une artériographie. Cette éventualité étant éliminée, il faut rechercher une tumeur hypophysaire, par l’étude du champ visuel et la radiographie de la selle turcique.
En cas de tumeur hypophysaire assez volumineuse, il est préférable de l’enlever chirurgicalement par voie transcrânienne ou par voie nasale transsphénoïdale. Sinon, on peut traiter d’abord le malade par des irradiations de l’hypophyse au moyen de la télécobaltothérapie comme dans l’acromégalie; ce traitement est rarement suffisant. On a eu recours également, avec des succès, à des implantations d’yttrium radio-actif dans la selle turcique. Souvent, on est amené à enlever chirurgicalement les surrénales hyperplasiques. Les surrénalectomies dites subtotales, comportant l’ablation d’une surrénale et des trois quarts de l’autre, sont habituellement suivies de récidives. Aussi doit-on se résoudre à la suppression complète des deux surrénales, d’emblée ou en deux temps. Cette intervention radicale permet la régression de tous les symptômes; mais le malade se trouve dès lors dans un état définitif d’insuffisance surrénale. Celle-ci est aisément compensée par des doses appropriées de cortisone et de désoxycorticostérone; et, finalement, cette situation est préférable à l’aggravation progressive de la maladie de Cushing, qui, en l’absence de traitement, conduit à la mort dans un délai de cinq à dix ans. Il est prudent de compléter la surrénalectomie par des irradiations de l’hypophyse, car la suppression des surrénales ne fournit qu’une guérison clinique, sans remédier à l’hyperactivité antéhypophysaire causale à laquelle elle donne au contraire un coup de fouet. Il n’est pas rare de voir une tumeur hypophysaire, restée jusqu’alors latente, se révéler après la surrénalectomie.
Il existe un traitement médicamenteux de la maladie de Cushing, utilisant des produits de synthèse comme l’Op’DDD ou l’aminoglutethimide qui, par une action antienzymatique, entravent la biosynthèse des hormones corticosurrénales. Il ne s’agit là que d’un traitement palliatif, d’action temporaire, et non dénué de toxicité. L’emploi de ces drogues sert donc surtout à préparer les malades à la surrénalectomie.
Panhypopituitarisme de l’adulte
Individualisé par M. Simmonds (1914), bien étudié par H. L. Sheehan (1938), ce syndrome traduit le déficit global des fonctions antéhypophysaires. Il résulte soit de certaines tumeurs hypophysaires, soit de divers processus vasculaires, infectieux ou inflammatoires détruisant la glande.
Les malades présentent une réduction générale de toutes les activités biologiques: asthénie profonde, physique et intellectuelle; frilosité; constipation; tendance à l’hypothermie; ralentissement du pouls; abaissement de la pression artérielle; sensations d’évanouissement (lipothymies) ou syncopes; aménorrhée chez la femme; perte de la libido chez l’homme. Leur faciès, pâle et un peu bouffi, prend un aspect vieillot, finement ridé. Les cheveux sont fins et secs; les poils pubiens et axillaires se raréfient, puis disparaissent. La pâleur ne tient pas seulement à un certain degré d’anémie, mais surtout à la réduction générale de la pigmentation.
Les examens biologiques montrent une baisse très importante du métabolisme basal; une tendance à l’hypoglycémie, avec sensibilité exagérée à l’insuline; un retard important de l’élimination de l’eau; une anémie modérée avec tendance à la mononucléose et éosinophilie discrète. Le défaut de stimulines hypophysaires entraîne une réduction considérable du fonctionnement des glandes qui sont sous leur dépendance. La thyroïde est la plus résistante, mais les corticosurrénales et les gonades voient leur sécrétion extrêmement réduite, ce qui se traduit par une disparition presque complète des 17-cétostéroïdes et des 17-hydroxycorticostéroïdes, la baisse du cortisol plasmatique, la disparition de toute activité gonadotrope dans les urines.
En principe, les gonades et les surrénales peuvent être réactivées par l’administration de gonadotrophine et de corticotrophine. Mais il est souvent nécessaire de répéter les épreuves pour obtenir une réponse significative. La stimulation de la thyroïde par la thyréostimuline est plus aisément obtenue.
Le panhypopituitarisme de l’adulte peut résulter du développement de tumeurs de l’hypophyse (adénomes chromophobes) ou de tumeurs juxta-hypophysaires (crâniopharyngiomes dérivés de restes embryonnaires de la poche de Rathke). Il est assez souvent la conséquence de nécroses de la glande par ischémie, notamment à la suite d’accouchements difficiles compliqués d’hémorragies (maladie de Sheehan). Il peut encore résulter de lésions diverses, d’origine infectieuse ou traumatique, de métastases cancéreuses ou de tumeurs cérébrales suprahypophysaires. Assez souvent son origine reste indéterminée.
Le panhypopituitarisme, indépendamment de sa cause, est compatible avec d’assez longues survies. La mort est généralement due à un « stress » intercurrent: infection, intoxication, troubles digestifs, traumatisme accidentel ou chirurgical, auxquels ces sujets offrent peu de résistance. L’asthénie fait alors place à la torpeur et au coma, dont une thérapeutique appropriée peut encore tirer les malades.
En dehors de tout facteur associé, une assez bonne condition physique peut être rétablie grâce à l’administration de cortisone, de petites doses d’extrait thyroïdien et éventuellement d’hormones sexuelles. En cas de coma, toutes les méthodes habituelles de réanimation doivent être mises en œuvre, en plus d’une hormonothérapie substitutive massive, surtout corticosurrénale.
Insuffisances antéhypophysaires à début prépubertaire
Installé avant la puberté, parfois congénitale, l’insuffisance antéhypophysaire comporte comme symptômes majeurs l’insuffisance de la croissance et la persistance d’un infantilisme génital après l’âge normal de la puberté.
De cette façon se trouve réalisé le tableau de l’infantilo-nanisme hypophysaire , qu’une étude clinique et biologique approfondie doit différencier d’autres variétés d’infantilonanismes.
Moins profond, ou bien d’installation plus tardive, l’hypopituitarisme peut réaliser le tableau moins sévère d’un infantilisme pur , qu’il importe de ne pas confondre avec les simples retards pubertaires, beaucoup plus fréquents et souvent familiaux.
Le syndrome adiposo-génital (J. Babinski, A. Fröhlich, 1901) associe le défaut de développement sexuel, avec ou sans insuffisance staturale, et une obésité importante. Mais celle-ci résulte de lésions hypothalamiques concomitantes, plutôt que de l’hypopituitarisme lui-même.
L’insuffisance hypophysaire prépubertaire peut résulter des mêmes causes que chez l’adulte, mais plus particulièrement de cranio-pharyngiomes (tumeurs de la poche de Rathke), de traumatismes obstétricaux, de séquelles de méningites, ou de simples défauts de développement de la région hypothalamo-hypophysaire. Elle s’accompagne assez souvent de troubles cérébraux associés: débilité mentale, épilepsie, troubles moteurs, etc.
Pour traiter l’insuffisance de la croissance, on utilise aujourd’hui la STH produite par génie génétique, de préférence à la somatotrophine extraite de tissus humains, laquelle a déterminé avant 1985 (date à laquelle sa purification a été perfectionnée) des accidents de contamination par le virus de la maladie de Creutzfeldt-Jakob. De petites doses d’extrait thyroïdien et, plus tard, l’administration de gonadotrophines, puis d’hormones sexuelles compensent les déficits correspondants.
Insuffisances antéhypophysaires dissociées
Avant la puberté, tout aussi bien que chez l’adulte, l’insuffisance antéhypophysaire peut se présenter sous un aspect dissocié, n’affectant qu’une ou deux fonctions de la glande, alors que les autres sont conservées, ou même augmentées.
Tel est par exemple le cas de l’eunuchoïdisme hypogonadotrophique , qui représente un défaut isolé d’hormones gonadotropes, avec conservation des fonctions somatotrope, corticotrope et thyréotrope. Le tableau est alors celui d’une insuffisance génitale pure, du type de l’eunuchoïdisme. Mais l’absence de gonadotrophines dans l’urine différencie aisément ce syndrome de l’eunuchoïdisme vrai (insuffisance primaire des gonades) qui comporte, après l’âge de la puberté, un taux anormalement élevé de gonadotrophines dans l’urine.
Dans ces cas, le testicule est, en principe, stimulable par l’administration de gonadotrophines. Toutefois ce traitement reste généralement insuffisant, ce qui oblige à recourir aux hormones sexuelles.
Ce syndrome semble résulter de lésions purement hypothalamiques. Il coexiste quelquefois avec un défaut d’odorat (anosmie), lié lui-même à un défaut de développement des lobes olfactifs du cerveau (syndrome de De Morsier).
On peut citer aussi, chez les femmes, nombre d’aménorrhées hypothalamiques , parfois liées à des lésions acquises, mais qui apparaissent fréquemment, après la puberté, chez des jeunes filles ou des jeunes femmes, comme la conséquence d’un dérèglement psychique primitif, et de nature purement « fonctionnelle ». L’aménorrhée peut être isolée, ou associée à des troubles du comportement alimentaire: perte ou diminution de l’appétit (anorexie), sensation de faim excessive et besoin d’absorber une grande quantité d’aliments (boulimie), causes d’amaigrissements sévères (comme dans l’« anorexie mentale ») ou d’engraissements insolites. Ces cas peuvent comporter la persistance d’une activité gonadotrope folliculo-stimulante, décelable dans les urines et dans le sang, et admettre essentiellement pour cause la suppression de l’activité lutéo-stimulante cyclique qui conditionne normalement l’ovulation et la menstruation. Dans les cas les plus graves d’anorexie mentale, avec amaigrissement très sévère, l’inanition peut à son tour entraîner un déficit antéhypophysaire plus ou moins global, au point que le syndrome a pu être confondu avec le panhypopituitarisme primitif, d’origine organique.
Une psychothérapie appropriée, éventuellement associée à un isolement hors du cadre de vie habituel et à des mesures diététiques, doit en principe permettre le retour spontané de la menstruation, sans aucune hormonothérapie. Il peut être utile de stimuler la sécrétion des gonadostimulines par l’administration de clomiphène, ou même de stimuler directement les ovaires par l’administration de gonadotrophines d’origine humaine, à la fois folliculo-stimulante et lutéo-stimulante.
3. Syndromes posthypophysaires
Syndrome par défaut d’ADH
On ne connaît pas de tableaux cliniques précis imputables à un défaut ou à un excès d’ocytocine. En revanche, le défaut d’hormone antidiurétique (ADH), réalise le syndrome connu sous le nom de diabète insipide . Les tubes rénaux ayant perdu le pouvoir de réabsorber la majeure partie de l’eau filtrée en très grande abondance par les glomérules, il en résulte une émission intense d’urine (polyurie) entraînant une soif excessive (polydipsie) compensatrice. Les malades urinent cinq à dix litres et plus par vingt-quatre heures et, pour survivre, doivent ingérer des quantités égales d’eau. Tourmentés sans cesse par la soif, ils sont obligés de se lever plusieurs fois par nuit pour boire et uriner. Si l’on réduit leurs boissons à un volume normal, ils perdent rapidement du poids, se déshydratent, surtout aux dépens du secteur intracellulaire de leur organisme, et pourraient mourir si l’on prolongeait trop l’épreuve. On constate objectivement une augmentation de la concentration du sodium dans le sang, et l’impossibilité de concentrer l’urine, dont la densité reste basse (inférieure à 1,005), et dont l’osmolarité demeure toujours inférieure à celle du sang, ce qui traduit une « clearance de l’eau libre » constamment positive quelle que soit la sévérité de la restriction hydrique.
Cette épreuve de la soif, limitée à douze ou vingt-quatre heures, est nécessaire pour distinguer le diabète insipide vrai, par carence permanente d’hormone antidiurétique, de certaines vésanies de la soif qualifiées de potomanies , le plus souvent de cause psychique, et où le fait primitif n’est pas la polyurie, mais la polydipsie.
Diverses épreuves permettent encore de différencier les deux états, ce qui n’est pas toujours facile. Dans le diabète insipide vrai, contrairement à ce que l’on observe chez les sujets normaux et chez les potomanes, la polyurie provoquée expérimentalement par l’ingestion d’une assez grande quantité d’eau n’est réduite ni par l’injection intraveineuse de nicotine (épreuve de Cates et Garrod), ni par la perfusion de sérum salé hypertonique (épreuve de Carter et Robbins), ni par la simple ingestion de sel (épreuve de Decourt). Il arrive toutefois que ces épreuves soient en défaut chez certains potomanes, dont les ingestions excessives et continues d’eau finissent par inhiber puissamment la sécrétion de l’hormone antidiurétique, au point que sa remise en route ne puisse se faire dans les conditions des épreuves. À ces faits convient le terme de « diabète induit et auto-entretenu » (Decourt). Il faut alors recourir à une méthode de déconditionnement, fondée sur la persuasion et sur un traitement fictif.
Le diabète insipide est souvent pur, installé après une infection, un traumatisme crânien, ou apparu sans cause décelable. Il peut s’associer à des troubles antéhypophysaires ou à des syndromes neurologiques, en rapport, par exemple, avec une tumeur suprahypophysaire ou avec toute autre altération pathologique affectant l’appareil hypothalamo-posthypophysaire.
Il doit être distingué des faux diabètes insipides néphrogéniques , le plus souvent congénitaux, qui réalisent un tableau identique, mais lié à une insensibilité des reins à l’action de l’hormone antidiurétique, laquelle est normalement sécrétée chez ces malades. Le diagnostic est assuré par l’épreuve de l’extrait posthypophysaire.
Dans le diabète insipide vrai, l’injection sous-cutanée d’extrait aqueux de posthypophyse corrige complètement la polyurie et la polydipsie, mais pour quelques heures seulement, ce qui rend le traitement peu praticable. L’hormone peut être administrée sous forme de poudre en prises nasales, mais ce procédé entraîne à la longue des altérations de la muqueuse qui risquent d’entraver le traitement. On possède heureusement des moyens indirects de réduire notablement la polyurie et la polydipsie: le régime déchloruré ou l’administration de médicaments déchlorurants et, mieux encore, la prise par voie buccale de produits de synthèse tels que le chlorpropamide, le tolbutamide, et surtout le clofibrate, qui exercent sur les reins une action assez comparable à celle de l’extrait posthypophysaire.
Syndromes par excès d’ADH
On ne connaît guère de syndromes liés à un excès d’hormone antidiurétique , du moins dans la pathologie proprement hypophysaire ou neurohypophysaire. De tels syndromes ont été postulés, plutôt que démontrés, par J. Bauer et par G. Zondek. J. Decourt a rapporté des observations qui paraissent y répondre. Il s’agissait de femmes qui présentaient des crises de rétention hydrique, associées à un dérèglement hypothalamique complexe, comportant des troubles des règles, des accès de boulimie avec engraissement et des troubles caractériels, et chez lesquelles les urines avaient un pouvoir antidiurétique quatre à six fois supérieur à celui que l’on constate chez les sujets normaux.
W. Schwartz et F. C. Bartter ont décrit en 1957 un syndrome de « sécrétion inappropriée d’hormone antidiurétique » chez des sujets porteurs de cancers non endocriniens, surtout de cancers bronchiques à petites cellules. Ce syndrome se caractérise par une diminution du sodium sanguin, associée à une hyperosmolarité urinaire. Il a pu être reproduit chez des sujets normaux soumis à la fois à une hyperhydratation et à la perfusion d’hormone antidiurétique. Il comporte bien l’excès d’un principe antidiurétique, analogue ou identique à la vasopressine, mais émané des cellules cancéreuses et non du complexe hypothalamo-posthypophysaire. On connaît d’ailleurs d’autres variétés de « syndromes paranéoplasiques » de mécanisme comparable, d’allure typiquement endocrinienne, mais liés de même à la production de substances « hormono-semblables » par certains cancers non endocriniens. Il existe, par exemple, un syndrome de Cushing reconnaissant une telle origine.
hypophyse [ ipɔfiz ] n. f.
• 1818; du gr. hupophusis « croissance en dessous »
♦ Organe neuroglandulaire ellipsoïde, situé à la base du crâne et rattaché au cerveau par la tige pituitaire (appelé aussi corps ou glande pituitaire). L'hypophyse, glande endocrine, sécrète plusieurs hormones qui agissent sur le fonctionnement d'autres glandes endocrines. ⇒ A. C. T. H., gonadotrope, mélanostimuline, ocytocine, prolactine, somatotrope.
● hypophyse nom féminin (grec hupophusis) Petite glande endocrine (sécrétant des hormones) reliée à la partie antérieure du cerveau. ● hypophyse (difficultés) nom féminin (grec hupophusis) Orthographe Attention à l'orthographe de ce mot difficile, qui prend deux h et deux y.
hypophyse
n. f. ANAT, PHYSIOL Glande endocrine logée dans la selle turcique (sous le cerveau) au-dessous de l'hypothalamus et qui, sécrétant les stimulines qui agissent sur les autres glandes endocrines, joue un rôle majeur dans la régulation des sécrétions hormonales. (L'hypophyse sécrète aussi un certain nombre d'autres hormones qui agissent en particulier sur la croissance [ hormone somatotrope, ou hormone de croissance ], sur la teneur du corps en eau et la teneur du sang en glucose.)
⇒HYPOPHYSE, subst. fém.
ANAT. Glande endocrine située à la base du cerveau et sécrétant de nombreuses hormones nécessaires au fonctionnement normal de l'organisme. L'hypophyse ou glande pituitaire, par son lobe antérieur, secrète [sic] des hormones excitant directement ou indirectement la croissance (MOUNIER, Traité caract., 1946, p. 169). L'hypophyse, petite glande grosse comme un pois et appendue à la base du cerveau (R. SCHWARTZ, Nouv. remèdes et mal. act., 1965, p. 36) :
• L'importance de l'hypophyse dans les régulations hormonales, a peu à peu destitué la thyroïde de son rang de glande privilégiée dans les relations entre endocrines et psychisme. L'hypophyse antérieure peut être actuellement considérée, selon l'expression de Cushing, comme un véritable cerveau endocrinien.
DELAY, Psychol. méd., 1953, p. 217.
— En appos. Glande hypophyse. La brièveté des pattes du basset tiendrait à une moindre activité de la glande hypophyse au moment de la pousse des membres (CUÉNOT, J. ROSTAND, Introd. génét., 1936, p. 30).
REM. Hypophysectomie, subst. fém. Ablation chirurgicale de l'hypophyse. L'hypophysectomie supprime la sensibilité des souris au cancer mammaire (P. MORAND, Confins vie, 1955, p. 153).
Prononc. : [ ]. Étymol. et Hist. 1818 (Dict. des sc. méd. ds Fr. mod. t. 37, p. 40, note 16). Adaptation du gr.
]. Étymol. et Hist. 1818 (Dict. des sc. méd. ds Fr. mod. t. 37, p. 40, note 16). Adaptation du gr. 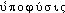 « naissance, ou croissance en dessous » composé de
« naissance, ou croissance en dessous » composé de  « sous » et
« sous » et 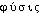 « formation ». Fréq. abs. littér. : 13.
« formation ». Fréq. abs. littér. : 13.
]. Étymol. et Hist. 1818 (Dict. des sc. méd. ds Fr. mod. t. 37, p. 40, note 16). Adaptation du gr. « naissance, ou croissance en dessous » composé de « sous » et « formation ». Fréq. abs. littér. : 13.
hypophyse [ipɔfiz] n. f.
ÉTYM. 1818, in T. L. F.; du grec hupophusis « croissance en dessous », de hupophuein « naître, croître », de hupo (→ Hypo-), et phuein « pousser ».
❖
♦ Biol. Organe neuro-glandulaire (glande endocrine) ellipsoïde, situé à la base du crâne (selle turcique) et rattaché au cerveau par la tige pituitaire. || Lobes antérieur, intermédiaire, inférieur de l'hypophyse. || L'hypophyse, « glande pituitaire » de l'ancienne médecine, est considérée comme un véritable « cerveau endocrinien ». || Sécrétions de l'hypophyse. → Gonade, cit. || L'hypophyse sécrète plusieurs hormones qui agissent sur le fonctionnement d'autres glandes endocrines. ⇒ Gonadotrope, mélanostimuline, ocytocine, prolactine, somatotrope.
1 L'hypophyse antérieure peut être actuellement considérée, selon l'expression de Cushing, comme un véritable cerveau endocrinien. Elle sécrète en effet une multitude d'hormones, les stimulines, qui tiennent sous leur dépendance la sécrétion de toutes les autres glandes endocrines; ovaires et testicules, thyroïde et parathyroïde, pancréas, cortico-surrénale et médullo-surrénale lui sont subordonnés (…) l'expression bien frappée qu'a employée Cushing est doublement justifiée : l'hypophyse est le cerveau endocrinien, non seulement parce qu'elle est la glande maîtresse mais parce qu'elle est la glande cérébrale. Sa situation à la base du cerveau, reliée à l'hypothalamus par la tige pituitaire, laissait présager d'étroites corrélations hypophyso-hypothalamiques qu'a confirmées l'étude histo-physiologique faite en particulier par Roussy et Mosinger et par Rémy Collin.
Jean Delay, la Psycho-physiologie humaine, p. 55-56.
2 L'hypophyse, par ses stimulines, règle le concert endocrinien; elle est le cerveau « végétatif » (…) « Là, écrivait Harvey Cushing, dans cette petite zone médiane et archaïque de la base du cerveau que pourrait cacher l'ongle du pouce, se dissimule le ressort essentiel de la vie instinctive et affective, que l'Homme s'est efforcé de recouvrir d'un manteau, d'une écorce (…) d'inhibitions ». Des réflexes hypophyso-sexuels peuvent avoir des origines : cutanée, olfactive, auditive, optique, psychique.
R. Kehl, les Glandes endocrines, p. 14.
❖
DÉR. Hypophysaire.
COMP. Antéhypophyse, hypophysectomie. — V. Hypophyso-.
Encyclopédie Universelle. 2012.